Hi there,
What could be yje reason for the error:
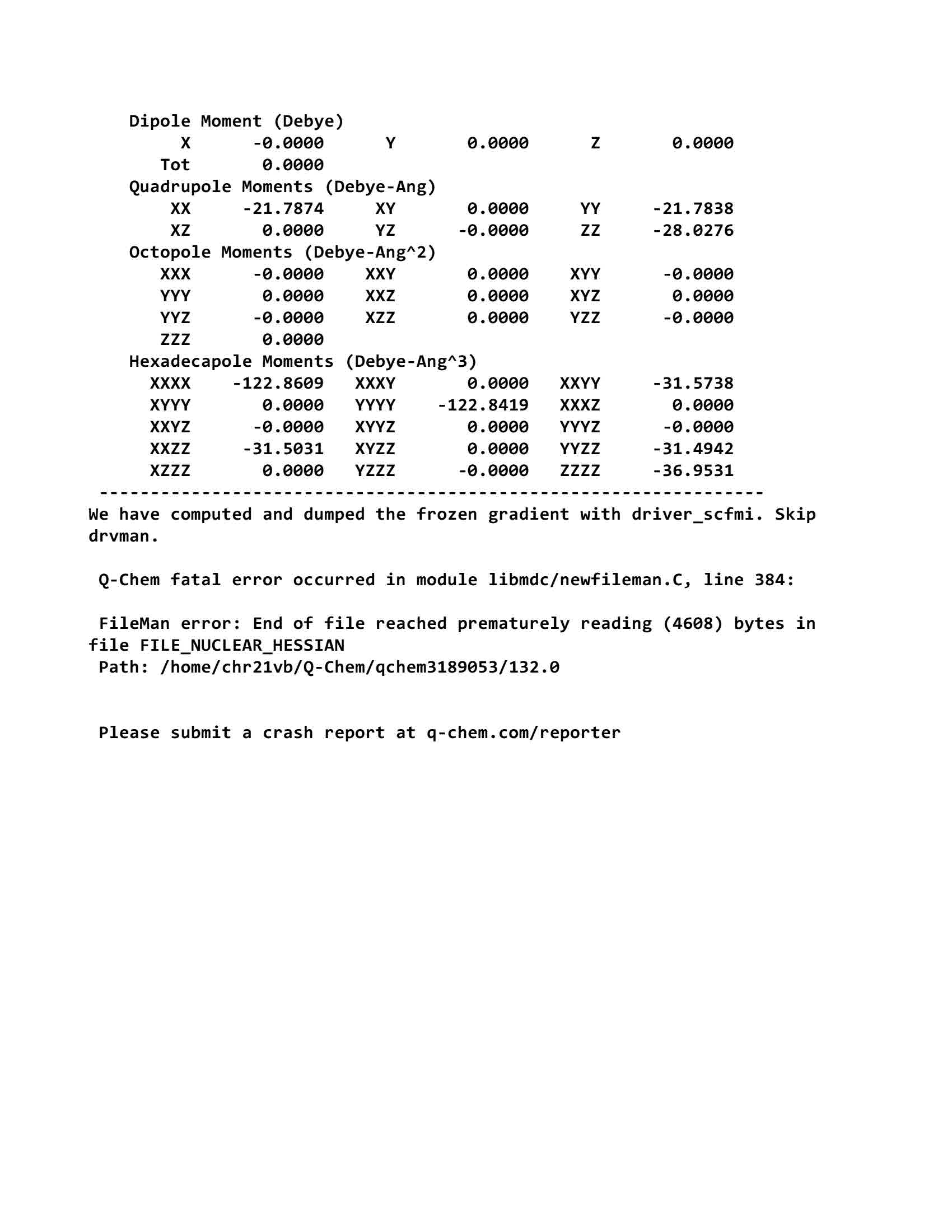
Q-Chem fatal error occurred in module libmdc/newfileman.C, line 384:
FileMan error: End of file reached prematurely reading (4608) bytes in file FILE_NUCLEAR_HESSIAN
Path: /home/Q-Chem/qchem3189053/132.0
$comment
CBD CAS FREQ 6-311++G(d,p)
$end
$molecule
0 1
C 0.7222339594 0.7222052838 0.0000000000
C -0.7222339594 0.7222052838 0.0000000000
C -0.7222339594 -0.7222052838 0.0000000000
C 0.7222339594 -0.7222052838 0.0000000000
H 1.4793367443 1.4792808434 0.0000000000
H -1.4793367443 1.4792808434 0.0000000000
H -1.4793367443 -1.4792808434 0.0000000000
H 1.4793367443 -1.4792808434 0.0000000000
$end
$rem
JOBTYPE = FREQ
EXCHANGE hf
UNRESTRICTED = FALSE ! For Closed-shell systems
BASIS 6-311++G(d,p)
SCF_CONVERGENCE = 8
POP_MULLIKEN = FALSE
SYMMETRY = FALSE
GEOM_OPT_MAX_CYCLES = 100
GEOM_OPT_PRINT = 0 !Hessian print out.
sym_ignore = TRUE !keyword is now correctly set to TRUE if NO_REORIENT=TRUE
GUI = 2 !Enabled generation of formatted checkpoint Formatted checkpoint (.fchk) files
mem_total = 60000
mem_static = 10000
RAMAN FALSE
MAX_SCF_CYCLES 400
THRESH 12 ! Should be at least three greater than SCF_CONVERGENCE =14 For coupled-cluster calculations Cutoff for neglect of two electron integrals
PRINT_ORBITALS = false
SCF_PRINT = 0 !breakdown of SCF electronic energy
SCF_FINAL_PRINT = 0 !
CAS_METHOD 2 ! 1 for CAS-CI, 2 for CASSCF (orbital optimization)
CAS_M_S 0 ! M_s value*2 The number of unpaired electrons desired in the CAS wavefunction
CAS_N_ELEC 4 ! N_elec Specifies the number of active electrons
CAS_N_ORB 4 ! N_orb Specifies the number of active orbitals
CAS_N_ROOTS 1 ! 1 ground state Specifies the number of electronic states to determine
CAS_THRESH 12 ! N for a threshold of 10^N. Specifies the threshold for matrix elements to be included in the CAS Hamiltonian
CAS_SAVE_NAT_ORBS FALSE ! Save the CAS natural orbitals in place of the reference orbitals
MAX_CASSCF_CYCLES 400 !Maximum number of orbital optimization cycles for CASSCF
CAS_USE_RI FALSE !Compute 2-electron integrals analytically. Analytic integrals are more accurate
CAS_DAVIDSON_TOL 5 ! Default. Specifies the tolerance for the Davidson solver used in CAS
CAS_DAVIDSON_MAXVECTORS 10 ! Default
CAS_SOLVER 1 ! 2=ASCI, 1=Olsen, 0=naive. 1 CAS-CI/CASSCF
TRUNC_CI_LEVEL 0 ! default
ASCI_DIAG 2 ! Specifies the diagonalization procedure. Use 2 for best trade-off of speed and memory usage. Arma Sparse=0, Davidson=1, Eigen Sparse=2
SCF_ALGORITHM diis_gdm !Pulay DIIS+then later switch to geometric direct minimizationAlgorithm used for converging the SCF.
!For systems with small HOMO-LUMO gaps and DIIS fails to converge, LS_DIIS could help
SCF_GUESS = CORE
$end
Well, please give the context (CASSCF ground state) of what you are trying to calculate along with the minimal input for which you see this error using the preformatted text option (</>). Please remove unnecessary keywords related to geometry optimization in your REM. You can try computing the frequencies with finite difference using the “IDERIV = 1” setting and verify the results.
There’s a lot going on here, as Kaushik mentioned. For one thing, your comment that UNRESTRICTED = FALSE ! For Closed-shell systems
is not corrected. What you’re actually doing there is turning on ROHF, which is probably not what you want for a closed-shell system.
The deeper problem, which may be a bug, is that this calculation appears to behave as if we have analytic Hessians for CASSCF, which will be news to me if true. There’s no message at the top about finite-difference Hessians.
I would do as Kaushik suggests and choose a minimal sample, set IDERIV explicitly and see what happens.
Thank you and Kaushik. I added the option “IDERIV = 1” and the job runs so far (still not finished) with no error messages.
I did not fully understand your note that my comment that UNRESTRICTED = FALSE ! For Closed-shell systems is not corrected. What I’ve found in the Q-chem manual is:
"UNRESTRICTED Controls the use of restricted or unrestricted orbitals.
FALSE Closed-shell systems.
TRUE Open-shell systems.
OPTIONS:
FALSE Constrain the spatial part of the alpha and beta orbitals to be the same.
TRUE Do not Constrain the spatial part of the alpha and beta orbitals.
RECOMMENDATION:
Use FALSE - Closed-shell systems unless ROHF is desired. "
I run CBD - closed shell system.
I would appreciate it very much if you could clarify my mistake. Thank you
I think that the bug you have found is that the CASSCF analytic Hessian is not available but for some reason Q-Chem does not automatically switch to finite-difference. I will report this.
Regarding UNRESTRICTED = FALSE: what you are getting in this case is a restricted open-shell calculation, see:
For a close-shell molecule, what you probably want is simply spin-restricted (i.e., all orbitals doubly occupied with equal levels for alpha and beta). That is the default if you simply omit the UNRESTRICTED keyword.
In this case, however, it appears that the ASCI (CASSCF) code defaults to using RO so even if you omit UNRESTRICTED, that is what you get. In case of SCF convergence problems you should consider preceding this job with a normal spin-restricted calculation and reading in those orbitals as a guess.
Finally, IDERIV = 1 solved the problem and the frequency job was finished successfully. So thank you again. Yes, I was a bit confused fo the situation that the lack of this option resulted the end of Hessian file reading error.
I’m trying to calculat the RC of CBD automerization, so you absolutelly right, I dial with the closed shell system. I personally like to use RHF. This usually simplifies MO analysis but one of my colleges suggested to use UHF for the reason that RHF implementation in the Q-chem is not quite good. Could it be correct? Since CBD is a biradical in the TS state, I had to use some multireference method, here the CASSCF. I calculated TS and the corresponded set of frequencies and now hope to continue with the IRC calculations. Thus, I’m just curious, whether CASSCF works well with IRC if at all?
I would highly appreciate for your comment. Thank you
There is nothing wrong with our RHF implementation, to the best of my knowledge, and for a closed-shell molecule this should be the same as ROHF. However, at a TS there is some argument to use a spin-unrestricted calculation since RHF dissociates to a much too high energy, which is “fixed” by breaking spin symmetry.
I would say that our CASSCF is probably not well tested for IRC calculations. I would be inclined to use a (unrestricted) DFT calculation to locate the TS, as a starting point. You can always put a higher-level calculation on top of that geomery.
Thank you very much for your comments. It is vey important to me your oipinion that CASSCF is probably not well tested for IRC calculations. To my knowledge DFT methods, unfortunately, are less sutable for the systems with multireference (about 0.5,0.5) nature as CBD at biradical region is. Thus, in my opinion, it would be hard to be based on the geometry, calculated that way. In this situation it would be probably sutable to use “stre” or even to vary the corresponded geometry parameres manually and to optimize the rest of the geometry? In my last calculations I was kinda surprised that Q-Chem could identify the point group differently, either the same geometry in the input is given as z-matrix or Chartesian. I also obtained the influence of the method of specifying the input geometry on the SCF convergence. Could you see any reason for that or probably I’ve missed something? Thank you very much!
If CASSCF works for your IRC calculation then so be it, what I meant is that this feature in Q-Chem is relatively new so there’s not a lot of community experience with it.
I’m not sure that I understand your question about point group symmetry.
Something else you could try if CASSCF proves to be too expensive is that we do have gradients for Yamaguchi approximate spin purification, see: https://manual.q-chem.com/latest/sect_OpSing.html
Also not a lot of community experience with this.