Wether CDFT is only constrained DFT calculation or replace it with xygjos ?
And what is the reason for this error after I tried to replace it?
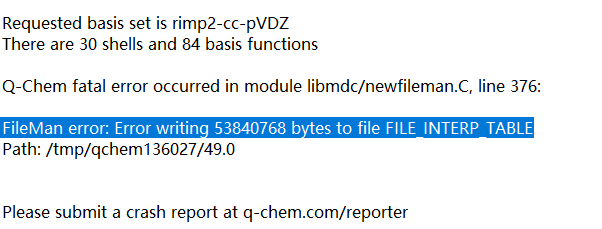
CDFT currently doesn’t support double-hybrid functionals such as XYGJOS. Standard semi-local or hybrid functionals would work.
Thanks for pointing this out, I have created a ticket to try to get a more informative error message in a future version of Q-Chem.
Please note that it’s usually necessary to provide at least a full input file to figure out these kinds of problems, unless someone like Yuezhi happens to know the answer (which wasn’t obvious to me).
Thank you for your answer. Can the ccsdt method be used in cdft? The other question is, is cdft equivalent to doing a single point or optimization?
Thank you for your answer. Does cdft support the coupled cluster method?
No, it is contrained Density Functional Theory.