Hi Q-Chem community,
We are currently investigating diabatic states based on bonding topology and are searching for appropriate methods for our system. For a given set of nuclear coordinates (this is all shown on the attached image), we wish to define diabatic states based on the connectivity (fragmentation). Previously, we have used another software to achieve this using cDFT, but Q-Chem seems to have more options.
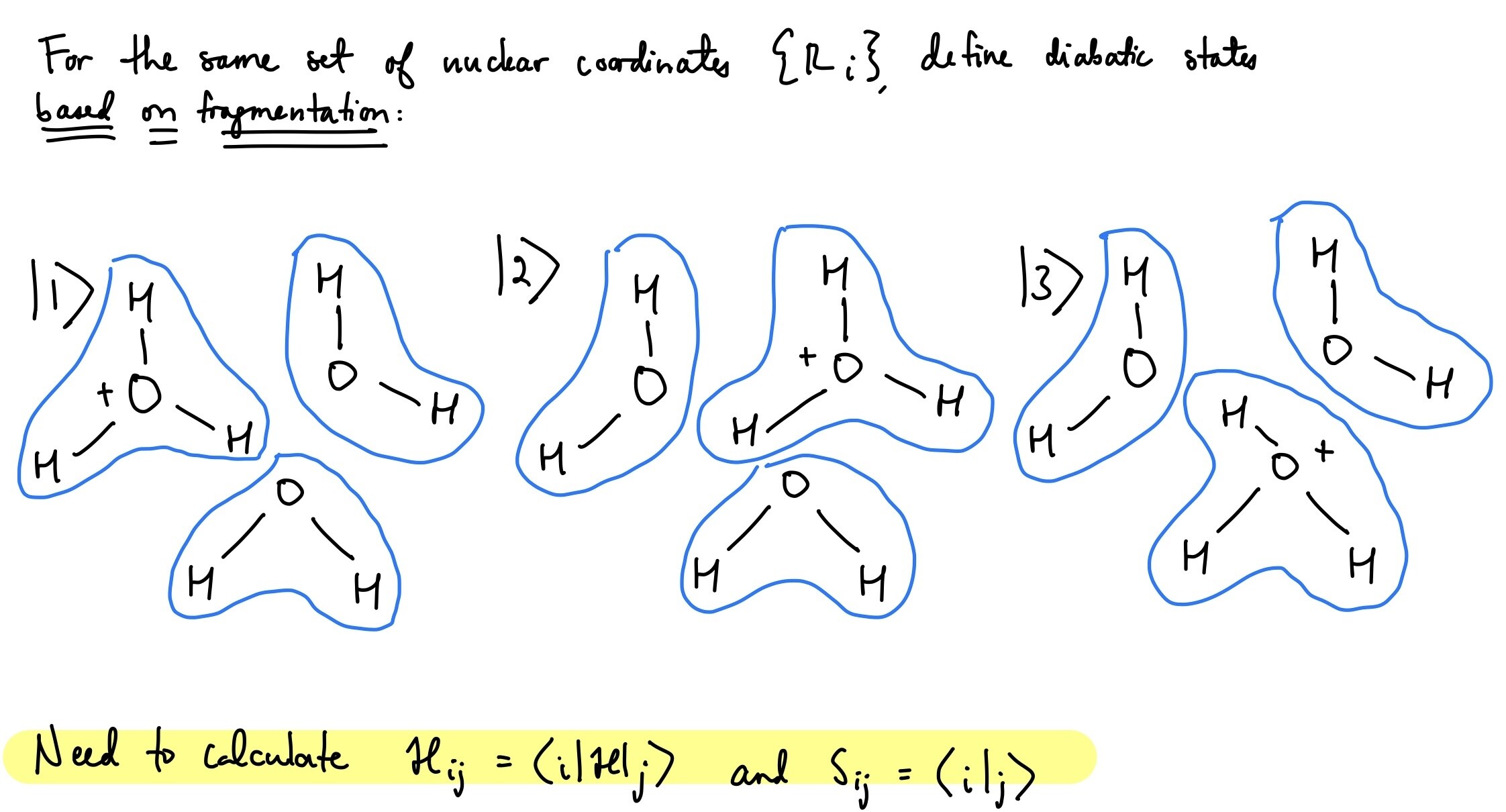
-
Is there a way to directly do fragment-based diabatization methods (POD, FODFT, ALMO) for more than two fragments/more than two states, and calculate the electronic couplings and overlaps? If not, is there a way to manually extract the output to generalize for more than two fragments/more than two states to obtain the full Hamiltonian and overlap matrices? The manual states that it is currently limited, but I wanted to ask if anyone had any thoughts or suggestions.
-
If the above is not feasible, could we use cDFT to fragmentize the system into more than two fragments, and calculate the couplings and overlaps between more than two states?
Thank you in advance!