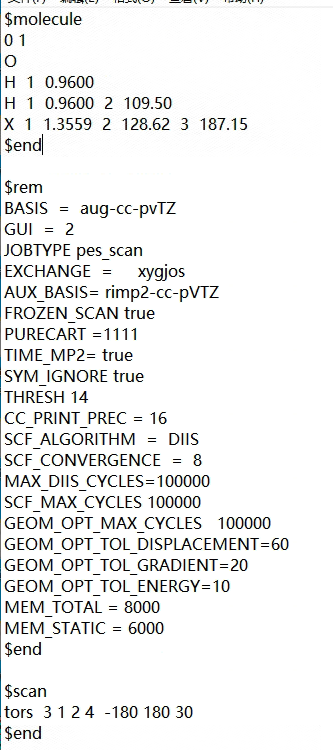
Dear Sirs and Madams, we try to set a dummy atom in order to do pes_scan in system. However, the output file is not show the transformation of energy.This is input file and output file.

Please let me know if there is any mistake.
Dear Sirs and Madams, we try to set a dummy atom in order to do pes_scan in system. However, the output file is not show the transformation of energy.This is input file and output file.
(a) What version of Q-Chem are you running? With the latest version (6.0.2), this job refuses to allow a constraint involving a dummy atom. It’s possible that was never supported but that earlier versions perhaps failed to check this (but I am not sure).
(b) What is it that you are trying to accomplish with this scan? Torsional angle scan in a triatomic molecule does not make sense to me.
We are using version 6.0.1.According to the discussion on the forum,we added FROZEN_SCAN true to the input file.However,the output file isn’t show change of energy.
It usually helps to look at the output file. If you do so, you will notice that the geometry is not changing (hence why the energy is the same at each point), and there is the following message:
The 3-1-2-4 torsion angle is not defined in the input z matrix
Therefore I ask again: What are you trying to accomplish with this calculation?
Thanks for you quention.In simple terms,we try to scan for energy.
As you said, that is not defined in the input Z matrix.So,should we change to internal coordinate or other.
I just don’t understand why you are trying to scan a torsional angle in a triatomic molecule.