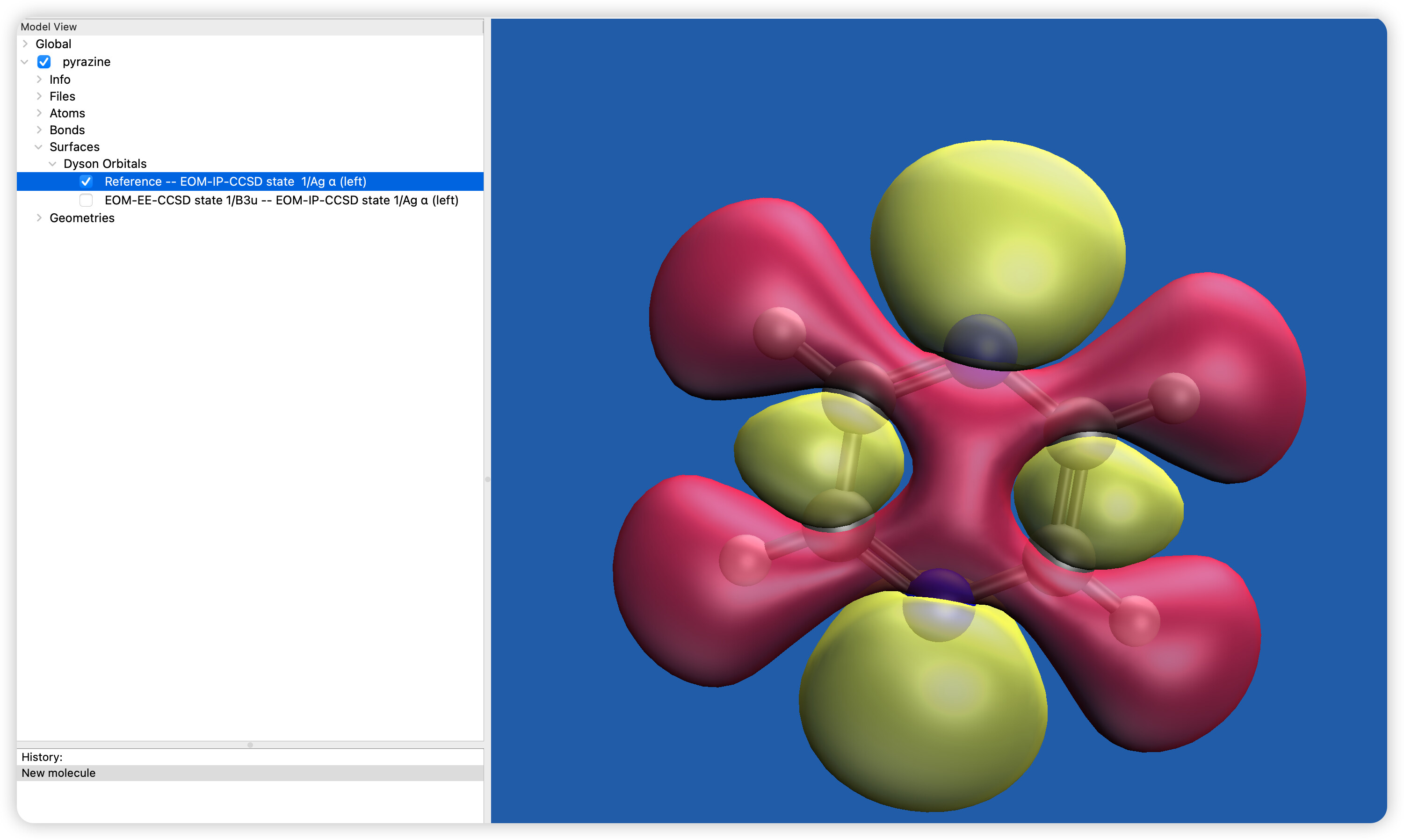
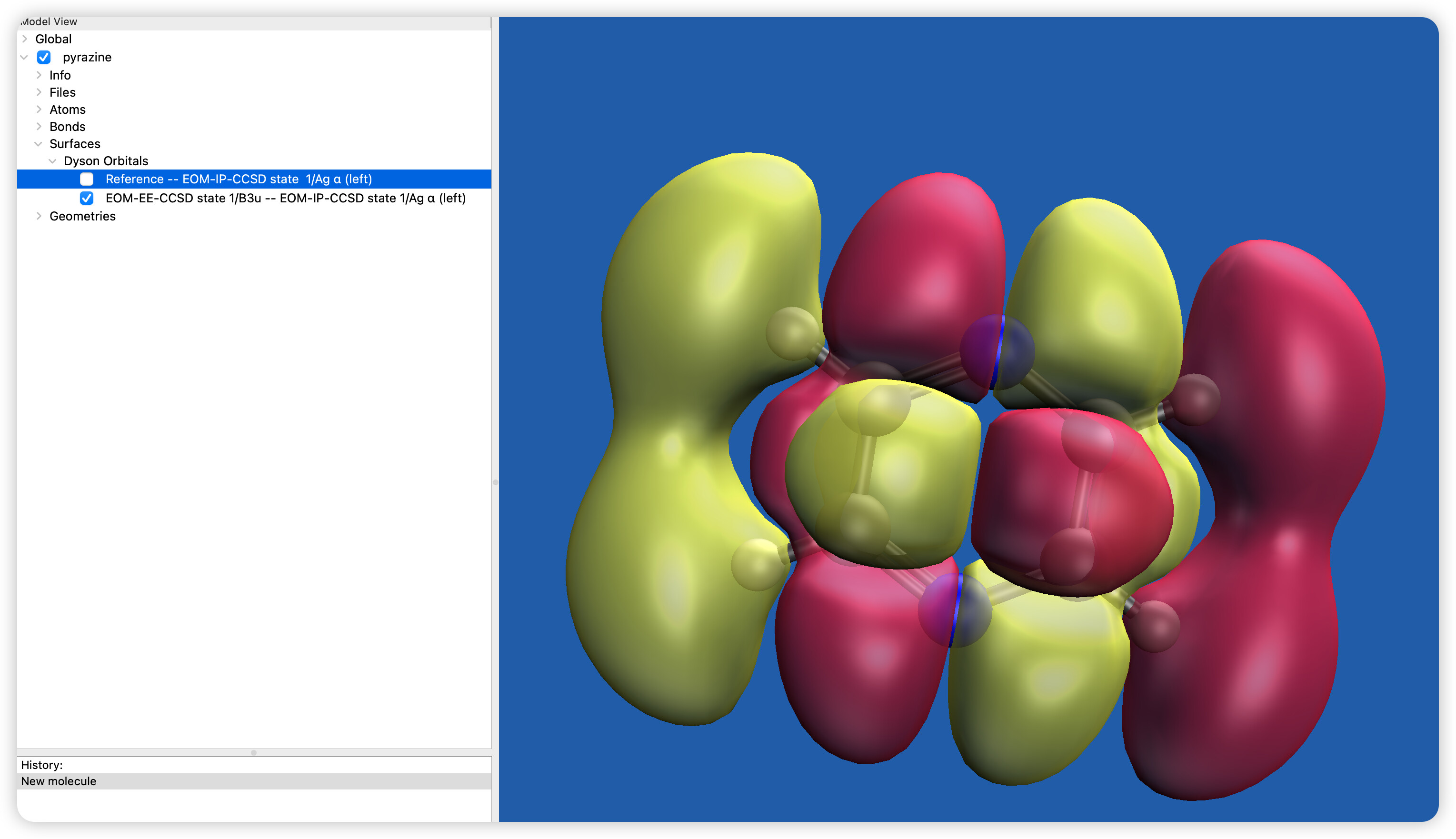
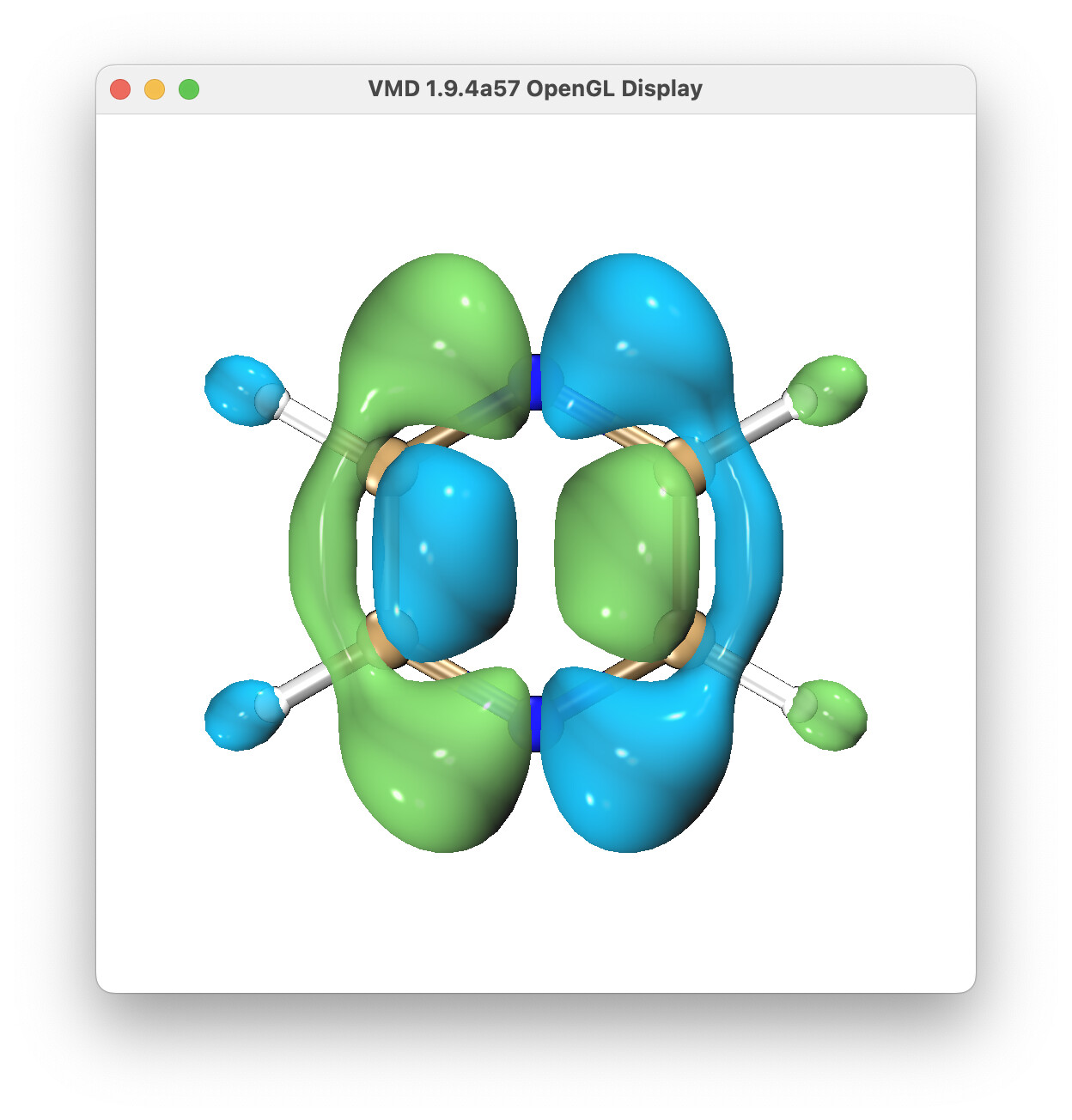
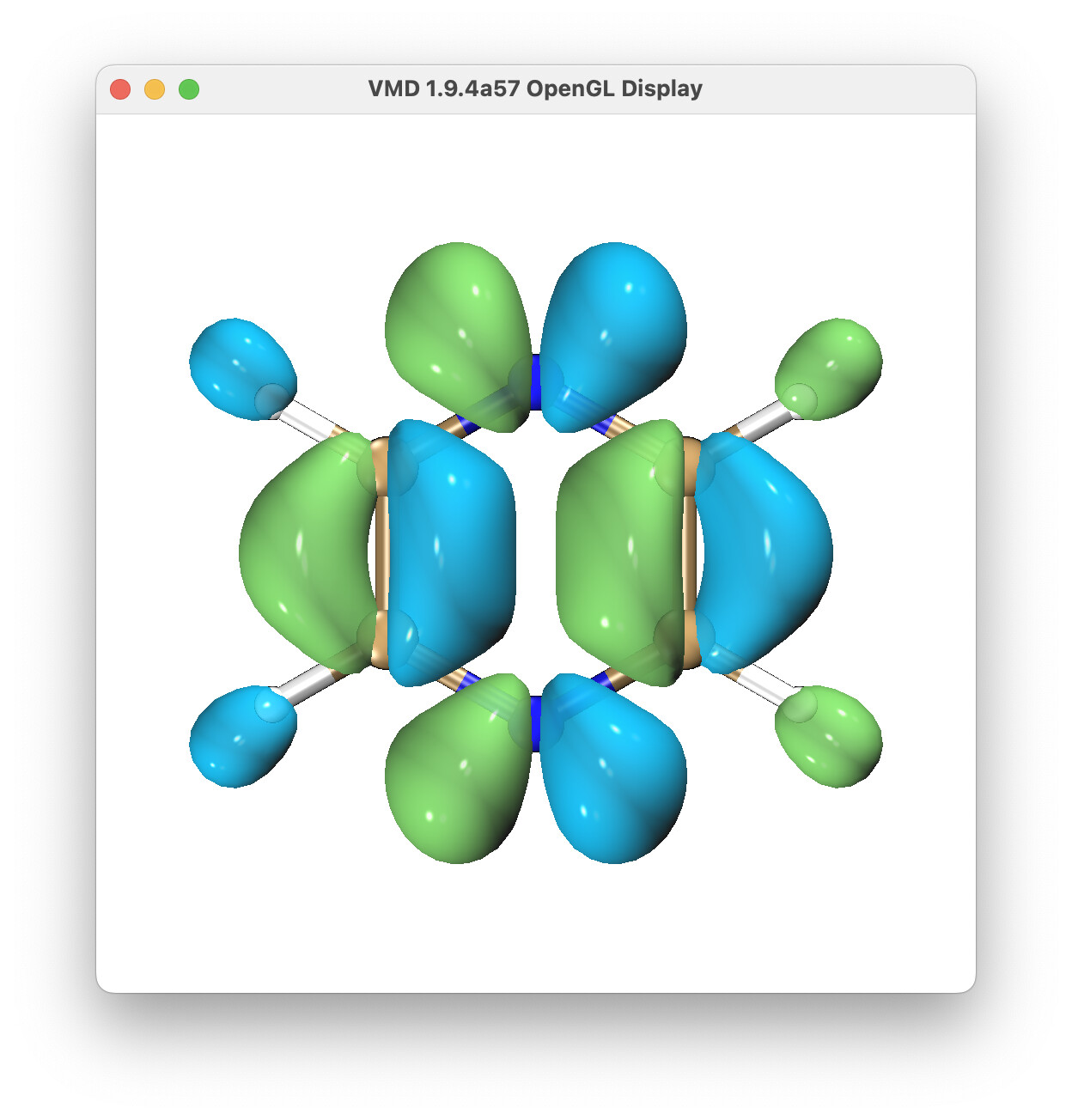
How to calculate the first excited state of pyrazine neutral molecule to its cation ground state of dyson orbital?
(the transition from pyrazine S1 (B3u) to its cation D0 (Ag), Ref[J. Chem. Theory Comput., 2021, 17, 5098−5105] )
Here is my script, how to modify it?
$molecule
0 1
C -1.207 0.695 0.000
C -1.207 -0.695 0.000
N 0.000 -1.388 0.000
C 1.207 -0.695 0.000
C 1.207 0.695 0.000
N 0.000 1.388 0.000
H -2.141 1.231 0.000
H -2.141 -1.231 0.000
H 2.141 -1.231 0.000
H 2.141 1.231 0.000
$end
$rem
METHOD eom-ccsd
BASIS 6-31G*
SCF_GUESS core
CCMAN2 true
EOM_IP_ALPHA [1,0,0,0,0,0,0,0] ; (Ag,B1g,B2g,B3g,Au,B1u,B2u,B3u)
CC_TRANS_PROP true necessary for Dyson orbitals job
CC_DO_DYSON true
PRINT_GENERAL_BASIS true will be required for ezDyson
MAKE_CUBE_FILES true
$end
You need to add additional keyword requesting to compute your desired excited state:
EOM_EE_SINGLETS [0,0,0,0,0,0,0,1] ; (Ag,B1g,B2g,B3g,Au,B1u,B2u,B3u).
You also need to specify for which transition you wish to compute transition property.
This can be done by:
CC_STATE_TO_OPT = [8,1] ! State 1 in irrep 8
Thank you very much for answering my question here. Do you mean to modify it to this? The output results seem to be very different from the first graph in Figure 3 of Ref [J. Chem. Theory Comput., 2021, 17, 5098−5105].
$molecule
0 1
C -1.207 0.695 0.000
C -1.207 -0.695 0.000
N 0.000 -1.388 0.000
C 1.207 -0.695 0.000
C 1.207 0.695 0.000
N 0.000 1.388 0.000
H -2.141 1.231 0.000
H -2.141 -1.231 0.000
H 2.141 -1.231 0.000
H 2.141 1.231 0.000
$end
$rem
METHOD eom-ccsd
BASIS 6-31G*
SCF_GUESS core
CCMAN2 true
EE_SINGLETS [0,0,0,0,0,0,0,1] ; (Ag,B1g,B2g,B3g,Au,B1u,B2u,B3u).
EOM_IP_ALPHA [1,0,0,0,0,0,0,0] ; (Ag,B1g,B2g,B3g,Au,B1u,B2u,B3u)
CC_STATE_TO_OPT [8,1]
CC_TRANS_PROP true necessary for Dyson orbitals job
CC_DO_DYSON true
PRINT_GENERAL_BASIS true will be required for ezDyson
MAKE_CUBE_FILES true
$end
Hello; it looks like your “reference - IP” Dyson orbital is consistent with the highest occupied Ag orbital appearing in Fig. 1 in the reference you cited. This suggests that you just have a different EOM-EE excited state than reported in that paper. This can be due to the different method, since they use CASPT2 that includes only pi* orbitals in the unoccupied part of the active space. Basis set can also make a difference. Try this instead to see if the second excited state matches:
EE_SINGLETS [0,0,0,0,0,0,0,2] ; (Ag,B1g,B2g,B3g,Au,B1u,B2u,B3u).
EOM_IP_ALPHA [1,0,0,0,0,0,0,0] ; (Ag,B1g,B2g,B3g,Au,B1u,B2u,B3u)
CC_STATE_TO_OPT [8,2]
Oh yeah this is because of difference in the symmetry conventions! Irrep changes depending on whether you define the principal rotation axis as the one perpendicular to the plane of the molecule or the one passing through most atoms. It’s always a good idea to check HF orbitals first. The orbital you want is actually B1u in Q-Chem, so you will need to use EE_SINGLETS=[0,0,0,0,0,1,0,0]. See here for more information: https://iopenshell.usc.edu/resources/howto/symmetry/