$molecule
1 1
1 1
Na -0.7887175826 -0.0886091306 0.2588198803
0 1
O -0.5906393086 1.5346995476 -1.5869257743
H -1.1113704331 2.1205579137 -2.1551066680
H -0.4298837521 0.7176564696 -2.1087343574
0 1
O 2.8660764950 -0.0864720623 0.6722670194
H 3.6056341945 0.0967128055 0.0742736086
H 2.3433136302 0.7404321677 0.7242858739
0 1
O 0.8821832751 -1.7172348931 0.2985826370
H 1.7472272915 -1.2199606449 0.4278910076
H 0.9759135647 -2.5444077090 0.7932700523
0 1
O -0.4263680548 -1.1233895876 -1.9320726012
H 0.2214272362 -1.6265229951 -1.3852224704
H -0.8169618579 -1.7590475301 -2.5487762482
0 1
O -2.6157929321 -0.2564556063 1.5449930728
H -2.6472810623 -0.4499798652 2.4945323131
H -3.5465285214 -0.1826214985 1.2838524030
0 1
O 0.7497613825 1.7054798099 0.7201377826
H 0.4705009282 2.0286702628 -0.1658344470
H 0.6773445217 2.4654764324 1.3174457756
$end
$rem
SYM_IGNORE true
METHOD B3LYP
BASIS cc-pVDZ
MANY_BODY_INT true
THRESH 14
SCF_CONVERGENCE 8
$end
$mbe
order 5
$end
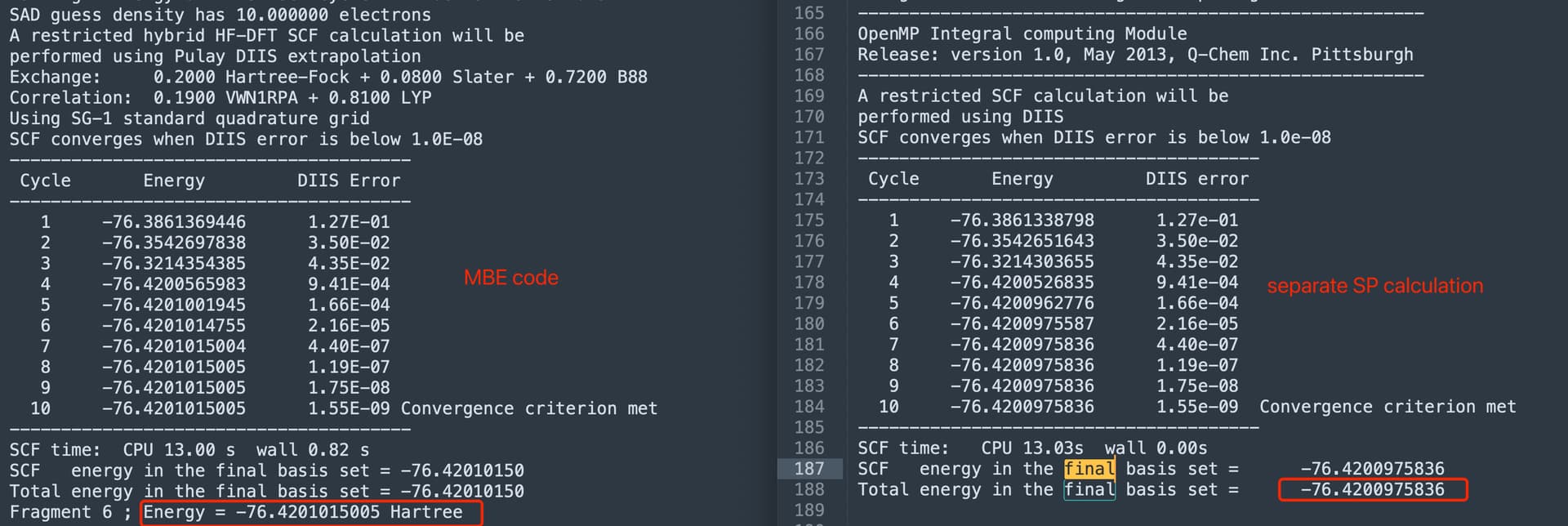
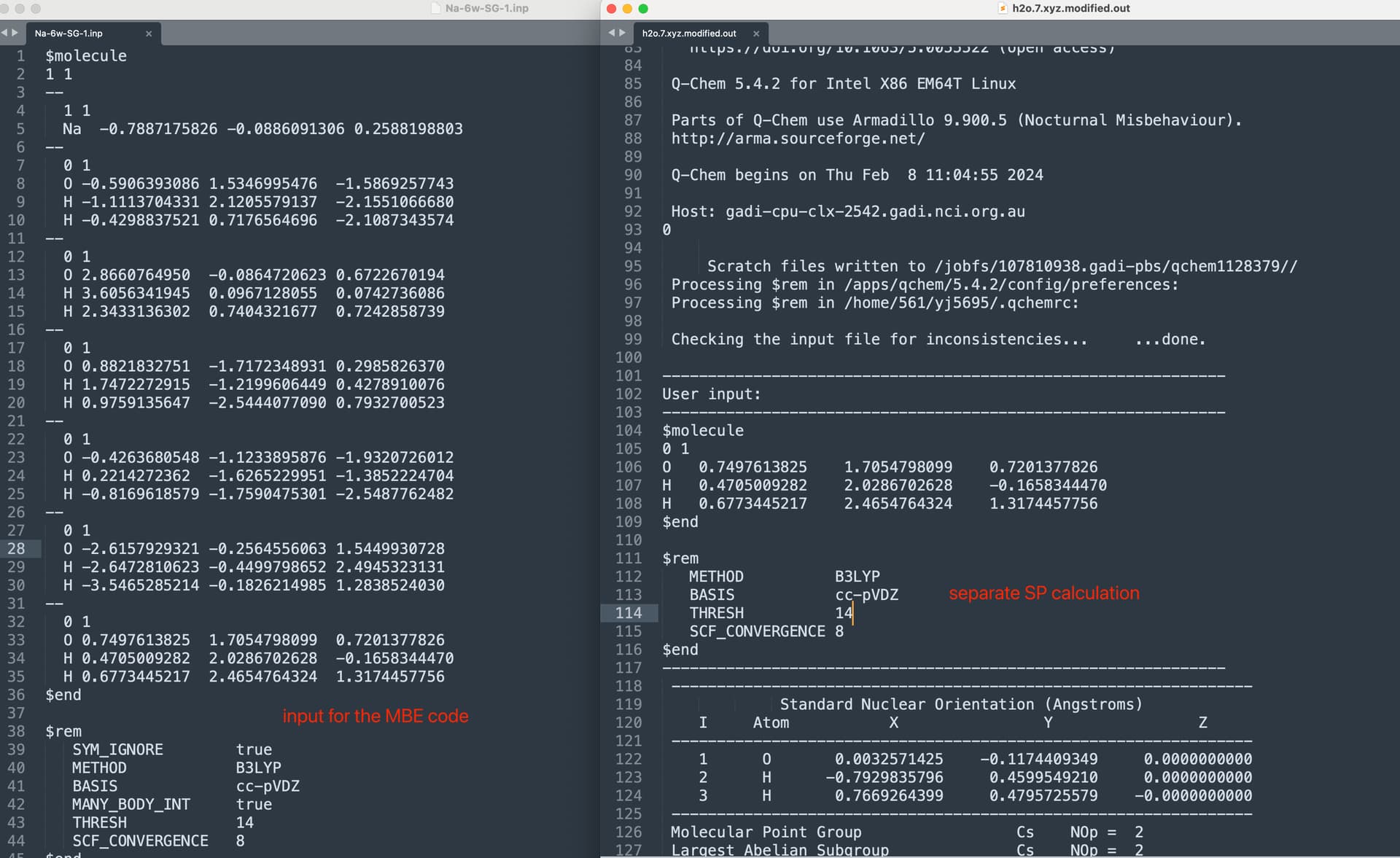
Above is the input file for the built-in MBE code. In order to check if there is an energy difference between the MBE code and the direct QM calculation, I extracted the last water monomer and performed a separate single point calculation (same level of theory, SCF and thresh). However, there is a slight energy difference between these two sets of calculations, see screenshot below. Does anyone know what is the reason for this?
Thanks in advance.