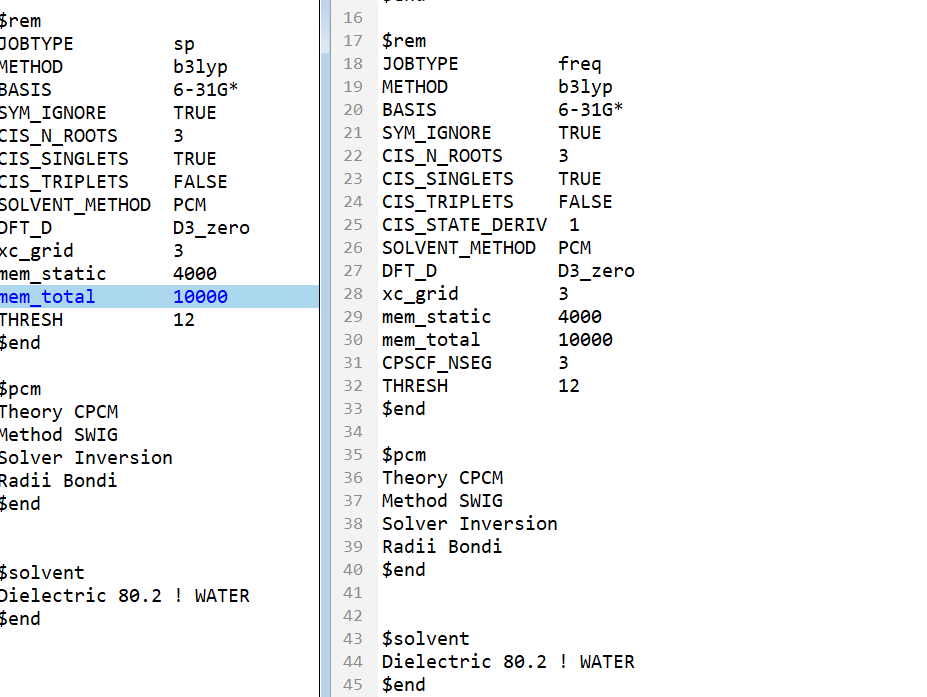
Hello! I was analyzing the frequency of the S1-state-benzene in water, and I also need to calculate the energy of S1-state-benzene. So I submitted two tasks for calculation. I found that the energy of S1-state-benzene in the two output files was inconsistent. What is the reason for this and which result should I use ?Here are my inputs.
here are my outputs
output1:
Excited state 1: excitation energy (eV) = 5.2907
Total energy for state 1: -232.05675062 au
Multiplicity: Singlet
Trans. Mom.: 0.0000 X -0.0001 Y -0.0000 Z
output2:
Excited state 1: excitation energy (eV) = 5.2801
Total energy for state 1: -232.05714100 au
Multiplicity: Singlet
Trans. Mom.: 0.0001 X -0.0002 Y -0.0000 Z
Could you post the full input file (using preformatted text button “</>”) and not paste an image for our convenience? Are you using the same version of Q-Chem for the two outputs? Also, are the SCF energies in the two calculations different? If so, set “SCF_CONVERGENCE = 8” in both your input files. The default SCF_CONVERGENCE is 5 for “JOBTYPE = sp” and 8 for “JOBTYPE = freq”. This difference in defaults in different job types could cause the excitation energies to differ slightly, so setting the same SCF_CONVERGENCE might resolve the issue.
That small of a different (0.01 eV) could easily be causes by the different convergence criteria, in my experience. By default, integral screening threshold and shell-pair drop tolerance are tied to the value of SCF_CONVERGENCE, which is different as Kaushik indicated.
Thank you for your suggestion. After setting the scf_comvergence of the “jobtype=sp” to 8, the calculation results of them are still inconsistent.
here are my inputs.
It seems that this has to do with the LR-PCM parts. If you set TDDFT_LR_PCM = FALSE, both jobs afford same results. I suspect this discrepancy is somehow triggered by the fact that the frequency job is computing a relaxed density on an excited state and the LR-PCM code may be modified in preparation for that, but without going into the code I am not sure.
If you do set TDDFT_LR_PCM = FALSE, you still get solvent effects in the TDDFT part at what I’ve called the “0th-order” approximation (solvent-polarized MOs and energy levels). https://doi.org/10.1002/wcms.1519