Hello everyone,
Is there a way to constrain the point-group symmetry (D₂h) during a 2-D scan (JOBTYPE=PES_SCAN), so that opposite C–C bonds remain equal each other at every grid point?
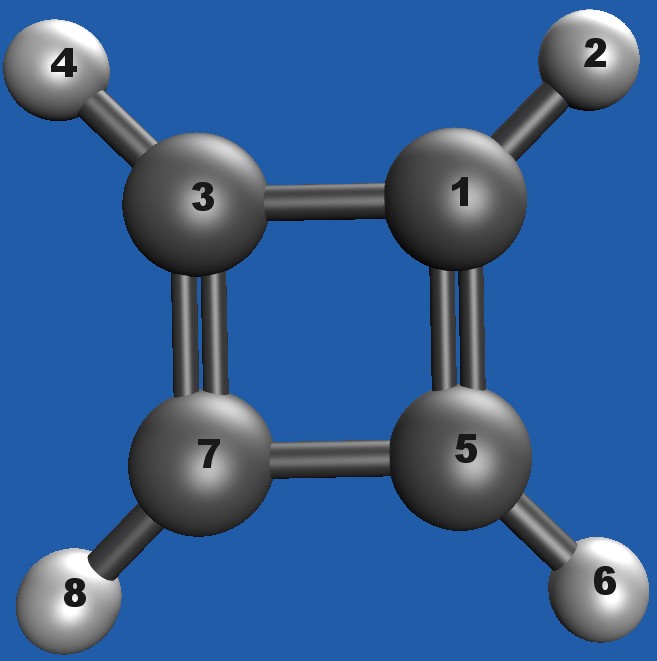
I’m scanning cyclobutadiene (CBD) with a symmetric Z-matrix. Opposite ring bonds are intended to stay equal: R(1–3) = R(5–7) and R(1–5) = R(3–7)
I run a 2-D frozen scan:
comment
CBD CAS pes_scan 6-311++G(d,p)
$end
$molecule
0 1
C
H 1 1.070965
C 1 R1 2 134.996727
H 3 1.070965 1 134.996727 2 -0.000000 0
C 1 R2 2 135.003273 3 -180.000000 0
H 5 1.070965 1 135.003273 2 -0.000000 0
C 5 R1 1 90.000000 2 180.000000 0
H 7 1.070965 5 134.996727 1 180.000000 0
R1=1.429784
R2=1.429784
$end
$rem
JOBTYPE = pes_scan !Find transition state
EXCHANGE hf
UNRESTRICTED = FALSE ! For Closed-shell systems
BASIS = 6-311++G(d,p)
SCF_CONVERGENCE = 8
POP_MULLIKEN = FALSE
SYMMETRY = TRUE
GEOM_OPT_MAX_CYCLES = 500
sym_ignore = FALSE
GUI = 2
mem_total = 20000
mem_static = 8190
MAX_SCF_CYCLES 500
THRESH 12
CAS_METHOD 2
CAS_M_S 0
CAS_N_ELEC 4
CAS_N_ORB 4
CAS_N_ROOTS 1
CAS_THRESH 12
MAX_CASSCF_CYCLES 500
CAS_USE_RI FALSE
CAS_DAVIDSON_TOL 5
CAS_DAVIDSON_MAXVECTORS 10
CAS_SOLVER 1
TRUNC_CI_LEVEL 0
ASCI_DIAG 2
SCF_ALGORITHM diis_gdm
FROZEN_SCAN TRUE
$end
$scan
stre 3 1 1.425 1.435 0.002
stre 5 1 1.425 1.435 0.002
$end
Q-Chem builds the full N×N grid (as expected), but the equality R(1–3)=R(5–7) is not maintained; e.g., As it is see in the attached distance matrix R(1–3)=1.435 while R(5–7) stays at its initial value (~1.429784).
Distance Matrix (Angstroms)
C ( 1) H ( 2) C ( 3) H ( 4) C ( 5) H ( 6)
H ( 2) 1.070965
C ( 3) 1.435000 2.319370
H ( 4) 2.319370 2.949487 1.070965
C ( 5) 1.435000 2.319424 2.029396 3.100361
H ( 6) 2.319424 2.949660 3.100361 4.171326 1.070965
C ( 7) 2.025712 3.096675 1.435009 2.321132 1.429784 2.314441
H ( 8) 3.096675 4.167640 2.317726 2.949664 2.314441 2.944271
C ( 7)
H ( 8) 1.070965
The question:
Can a 2-D PES_SCAN enforce either point-group symmetry*or equality constraints (e.g., R13=R57, R15=R37) at every grid point?
Thank you very much!