Hello everyone,
I am trying to use FED method to calculate coupling.
When calculate C2h4 dimer it worked. the input file is followed.
$molecule
0 1
C 0 0 0.665547
C 0 0 -0.665547
H 0 0.923577 1.23971
H 0 -0.923577 1.23971
H 0 -0.923577 -1.23971
H 0 0.923577 -1.23971
C 3.5 0 0.665547
C 3.5 0 -0.665547
H 3.5 0.923577 1.23971
H 3.5 -0.923577 1.23971
H 3.5 -0.923577 -1.23971
H 3.5 0.923577 -1.23971
$end
$rem
JOBTYPE SP
BASIS DZ*
CORRELATION RASCI
UNRESTRICTED FALSE
RAS_ROOTS 5
RAS_ACT 4
RAS_ELEC_ALPHA 2
RAS_ELEC_BETA 2
RAS_OCC 14
STS_FED TRUE
STS_ACCEPTOR 1-6
STS_DONOR 7-12
RAS_SPIN_MULT 1
MEM_TOTAL = 40000
MEM_STATIC = 10000
$end
But when calculate coupling between benzene, the error occurred.
The input file is
$molecule
0 1
C -1.69900000 1.56000000 -2.73900000
C -0.92800000 0.22700000 -2.73900000
C -1.69800000 -1.10600000 -2.73900000
C -3.23800000 -1.10400000 -2.73800000
C -4.01000000 0.22800000 -2.73800000
C -3.24000000 1.56100000 -2.73800000
H -1.12900000 2.54700000 -2.73800000
H 0.21200000 0.22800000 -2.73800000
H -1.12900000 -2.09300000 -2.73800000
H -3.80800000 -2.09200000 -2.73700000
H -5.15000000 0.22800000 -2.73600000
H -3.81000000 2.54800000 -2.73600000
C -3.96500000 0.24800000 0.23900000
C -3.19400000 -1.08500000 0.24000000
C -1.65400000 -1.08600000 0.23900000
C -0.88300000 0.24600000 0.23900000
C -1.65400000 1.57900000 0.23900000
C -3.19500000 1.58000000 0.23900000
H -5.10500000 0.24800000 0.23800000
H -3.76400000 -2.07200000 0.23800000
H -1.08500000 -2.07400000 0.23800000
H 0.25700000 0.24700000 0.23800000
H -1.08400000 2.56600000 0.23800000
H -3.76500000 2.56700000 0.23800000
$end
$rem
JOBTYPE SP
BASIS DZ*
CORRELATION RASCI
UNRESTRICTED FALSE
RAS_ROOTS 5
RAS_ACT 4
RAS_ELEC_ALPHA 2
RAS_ELEC_BETA 2
RAS_OCC 40
STS_FED TRUE
STS_ACCEPTOR 1-12
STS_DONOR 13-24
RAS_SPIN_MULT 1
MEM_TOTAL = 40000
MEM_STATIC = 10000
$end
and there is the error :
26 4 1 0.000004 0.000002
27 4 1 0.000004 0.000002
28 4 1 0.000004 0.000002
29 4 1 0.000003 0.000001
30 4 1 0.000003 0.000001
Q-Chem fatal error occurred in module 0, line 124:
MaxIt Reached in the diagonalization.
How can i fix it?
Use SET_ITER to increase the maximum number of Davidson iterations, as described in the manual.
https://manual.q-chem.com/6.0/Ch7.S12.SS1.html
hello, there is another error.
I want to calculate the triplet annihilation coulpling like <tt|H|s0s1> in dimer. I think if i get the optimized coordinate and wave function of two dimer state, and use fed method, the problem will solved. So I just tried <s0s1|H|s1s0> as an example using the pentacene dimer coordinate in
jz5b00437_si_002.pdf (acs.org)
the input file is
$molecule
0 1
C 1.27422619 -0.17599163 6.01483142
C 0.02892631 0.48552813 6.13157795
C -0.70441729 0.77801275 5.01145565
C -0.23516954 0.42697493 3.72115348
C -0.96866018 0.71548962 2.55146981
C -0.50831325 0.37108245 1.28530027
C -1.23273708 0.65484623 0.11556877
C -0.77123554 0.30863078 -1.16534292
C -1.49139597 0.59132472 -2.32084021
C -1.02099804 0.24031788 -3.60338811
C -1.75500870 0.52846683 -4.78088513
C -1.27422619 0.17599163 -6.01483142
C -0.02892631 -0.48552813 -6.13157795
C 0.70441729 -0.77801275 -5.01145565
C 0.23516954 -0.42697493 -3.72115348
C 0.96866018 -0.71548962 -2.55146981
C 0.50831325 -0.37108245 -1.28530027
C 1.23273708 -0.65484623 -0.11556877
C 0.77123554 -0.30863078 1.16534292
C 1.49139597 -0.59132472 2.32084021
C 1.02099804 -0.24031788 3.60338811
C 1.75500870 -0.52846683 4.78088513
H 1.84262471 -0.40130938 6.89992213
H -0.34012566 0.75819646 7.10455439
H -1.65166959 1.28139654 5.10259755
H -1.91611727 1.21918403 2.64510269
H -2.18070850 1.15842118 0.20444084
H -2.43987468 1.09477646 -2.23672961
H -2.70275752 1.03173269 -4.69437118
H -1.84262471 0.40130938 -6.89992213
H 0.34012566 -0.75819646 -7.10455439
H 1.65166959 -1.28139654 -5.10259755
H 1.91611727 -1.21918403 -2.64510269
H 2.18070850 -1.15842118 -0.20444084
H 2.43987468 -1.09477646 2.23672961
H 2.70275752 -1.03173269 4.69437118
C -4.67056507 -0.18506037 5.98840220
C -5.94763775 0.49331465 6.10812821
C -6.67418519 0.78452690 5.01611687
C -6.20621674 0.43116395 3.68898703
C -6.92840463 0.71743761 2.56444487
C -6.46043624 0.36657455 1.26793118
C -7.18885824 0.65382612 0.11539339
C -6.71978487 0.30497377 -1.14941001
C -7.45386033 0.59263077 -2.33323167
C -6.98494960 0.24619842 -3.56943974
C -7.72552911 0.53481048 -4.78326779
C -7.24543493 0.18506037 -5.98840220
C -5.96836225 -0.49331465 -6.10812821
C -5.24181481 -0.78452690 -5.01611687
C -5.70978326 -0.43116395 -3.68898703
C -4.98759537 -0.71743761 -2.56444487
C -5.45556376 -0.36657455 -1.26793118
C -4.72714176 -0.65382612 -0.11539339
C -5.19621513 -0.30497377 1.14941001
C -4.46213967 -0.59263077 2.33323167
C -4.93105040 -0.24619842 3.56943974
C -4.19047089 -0.53481048 4.78326779
H -4.11184469 -0.40466543 6.88131132
H -6.30554395 0.76061733 7.08697140
H -7.62149012 1.28782093 5.10603040
H -7.87644956 1.22096519 2.65247405
H -8.13676267 1.15734851 0.20425976
H -8.40172461 1.09620126 -2.24351870
H -8.67306844 1.03804884 -4.69553901
H -7.80415531 0.40466543 -6.88131132
H -5.61045605 -0.76061733 -7.08697140
H -4.29450988 -1.28782093 -5.10603040
H -4.03955044 -1.22096519 -2.65247405
H -3.77923733 -1.15734851 -0.20425976
H -3.51427539 -1.09620126 2.24351870
H -3.24293156 -1.03804884 4.69553901
C 1.28743493 -0.18506037 5.98840220
C 0.01036225 0.49331465 6.10812821
C -0.71618519 0.78452690 5.01611687
C -0.24821674 0.43116395 3.68898703
C -0.97040463 0.71743761 2.56444487
C -0.50243624 0.36657455 1.26793118
C -1.23085824 0.65382612 0.11539339
C -0.76178487 0.30497377 -1.14941001
C -1.49586033 0.59263077 -2.33323167
C -1.02694960 0.24619842 -3.56943974
C -1.76752911 0.53481048 -4.78326779
C -1.28743493 0.18506037 -5.98840220
C -0.01036225 -0.49331465 -6.10812821
C 0.71618519 -0.78452690 -5.01611687
C 0.24821674 -0.43116395 -3.68898703
C 0.97040463 -0.71743761 -2.56444487
C 0.50243624 -0.36657455 -1.26793118
C 1.23085824 -0.65382612 -0.11539339
C 0.76178487 -0.30497377 1.14941001
C 1.49586033 -0.59263077 2.33323167
C 1.02694960 -0.24619842 3.56943974
C 1.76752911 -0.53481048 4.78326779
H 1.84615531 -0.40466543 6.88131132
H -0.34754395 0.76061733 7.08697140
H -1.66349012 1.28782093 5.10603040
H -1.91844956 1.22096519 2.65247405
H -2.17876267 1.15734851 0.20425976
H -2.44372461 1.09620126 -2.24351870
H -2.71506844 1.03804884 -4.69553901
H -1.84615531 0.40466543 -6.88131132
H 0.34754395 -0.76061733 -7.08697140
H 1.66349012 -1.28782093 -5.10603040
H 1.91844956 -1.22096519 -2.65247405
H 2.17876267 -1.15734851 -0.20425976
H 2.44372461 -1.09620126 2.24351870
H 2.71506844 -1.03804884 4.69553901
C -4.68377381 -0.17599163 6.01483142
C -5.92907369 0.48552813 6.13157795
C -6.66241729 0.77801275 5.01145565
C -6.19316954 0.42697493 3.72115348
C -6.92666018 0.71548962 2.55146981
C -6.46631325 0.37108245 1.28530027
C -7.19073708 0.65484623 0.11556877
C -6.72923554 0.30863078 -1.16534292
C -7.44939597 0.59132472 -2.32084021
C -6.97899804 0.24031788 -3.60338811
C -7.71300870 0.52846683 -4.78088513
C -7.23222619 0.17599163 -6.01483142
C -5.98692631 -0.48552813 -6.13157795
C -5.25358271 -0.77801275 -5.01145565
C -5.72283046 -0.42697493 -3.72115348
C -4.98933982 -0.71548962 -2.55146981
C -5.44968675 -0.37108245 -1.28530027
C -4.72526292 -0.65484623 -0.11556877
C -5.18676446 -0.30863078 1.16534292
C -4.46660403 -0.59132472 2.32084021
C -4.93700196 -0.24031788 3.60338811
C -4.20299130 -0.52846683 4.78088513
H -4.11537529 -0.40130938 6.89992213
H -6.29812566 0.75819646 7.10455439
H -7.60966959 1.28139654 5.10259755
H -7.87411727 1.21918403 2.64510269
H -8.13870850 1.15842118 0.20444084
H -8.39787468 1.09477646 -2.23672961
H -8.66075752 1.03173269 -4.69437118
H -7.80062471 0.40130938 -6.89992213
H -5.61787434 -0.75819646 -7.10455439
H -4.30633041 -1.28139654 -5.10259755
H -4.04188273 -1.21918403 -2.64510269
H -3.77729150 -1.15842118 -0.20444084
H -3.51812532 -1.09477646 2.23672961
H -3.25524248 -1.03173269 4.69437118
$end
$rem
JOBTYPE SP
BASIS DZ*
CORRELATION RASCI
UNRESTRICTED FALSE
RAS_ROOTS 14
RAS_ACT 4
RAS_ELEC_ALPHA 2
RAS_ELEC_BETA 2
RAS_OCC 290
STS_FED TRUE
STS_ACCEPTOR 1-72
STS_DONOR 73-144
set_iter 5000
RAS_SPIN_MULT 1
$end
and here is the error.
“Input geometry has atoms that are too close for a meaningful calculation.
Correct atom coordinates and resubmit the job.”
How can I fix it?
Thank you!
don’t you need to define fragments for an FED job?
Sorry I don’t get the point, does it mean the molecule part like
"
$molecule
0 1
0 1
…
0 1
…
end
"
or using the key words like ras_frag_sets?
because the manual about FED is like
$molecule
0 1
C 0.670518 0.000000 0.000000
H 1.241372 0.927754 0.000000
H 1.241372 -0.927754 0.000000
C -0.670518 0.000000 0.000000
H -1.241372 -0.927754 0.000000
H -1.241372 0.927754 0.000000
C 0.774635 0.000000 4.000000
H 1.323105 0.936763 4.000000
H 1.323105 -0.936763 4.000000
C -0.774635 0.000000 4.000000
H -1.323105 -0.936763 4.000000
H -1.323105 0.936763 4.000000
$end
$rem
JOBTYPE SP
BASIS DZ*
CORRELATION RASCI
UNRESTRICTED FALSE
RAS_ROOTS 5
RAS_ACT 4
RAS_ELEC_ALPHA 2
RAS_ELEC_BETA 2
RAS_OCC 14
STS_FED TRUE
STS_ACCEPTOR 1-6
STS_DONOR 7-12
RAS_SPIN_MULT 1
$end
and I think the STS_ACCEPTOR and STS_DONOR can be the fragments part.
Do you mean I need more key words to make the dimer states be TT and S0S1?
but there are several states like the following, there are 6 TT states and 2 S0S1 states.
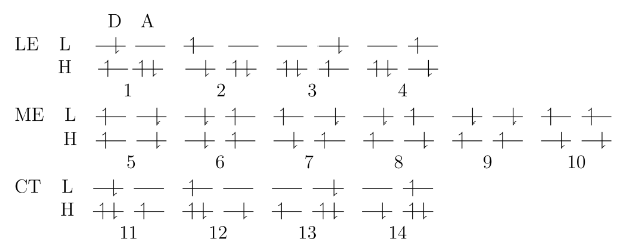
Looking at the examples in the manual, it looks like I was wrong about needing to specify the fragments in the $molecule section. (Other types of coupling calculations require this, the FED code must figure it out.) However, it seems like there’s something funny with your coordinates, because when I visualize the structure in IQmol most of the bonds are missing, meaning the atoms are unusually far apart. Could it be that you grabbed a geometry in Bohr but have assumed it was in Angstroms?
Thanks for answering me so fast, just let me explian the coordinates.
There are 144 atoms in the molecule parts. But the first 72 atoms and the other 72 atoms are nearly the same structure. The first 72 atoms represent a pentacene dimer optimized in S0S1 state, while the other 72 atoms represent the same pentacene dimer optimized in S1S0 state, so the distance between these two groups are really small. But I think I should concern the dimer as a whole part, so I write the coordinate in this way so that I can write the STS_DONOR and STS_ACCEPTOR as 1-72 and 73-144.
I know this may be wrong, but I can’t figure out other way to calculate the triplet annihilation/singlet fission couplings in dimer.
I don’t know how to do what you want but Q-Chem will definitely not let you use a structure that has (nearly) overlapping atoms, so that is the source of the error message that you see. The problem is that the nuclear-nuclear repulsion term goes to infinity and that messes with everything, hard to get a finite answer.