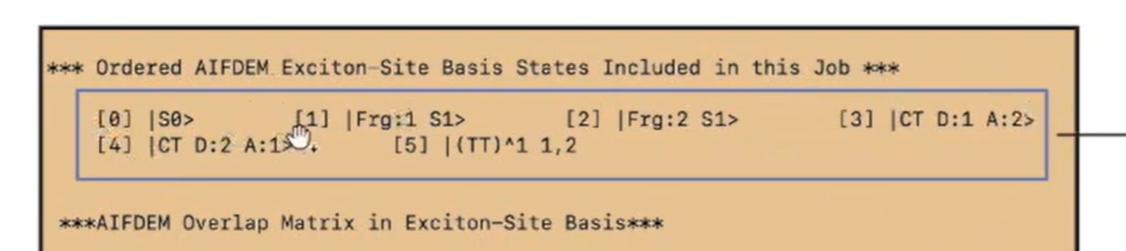
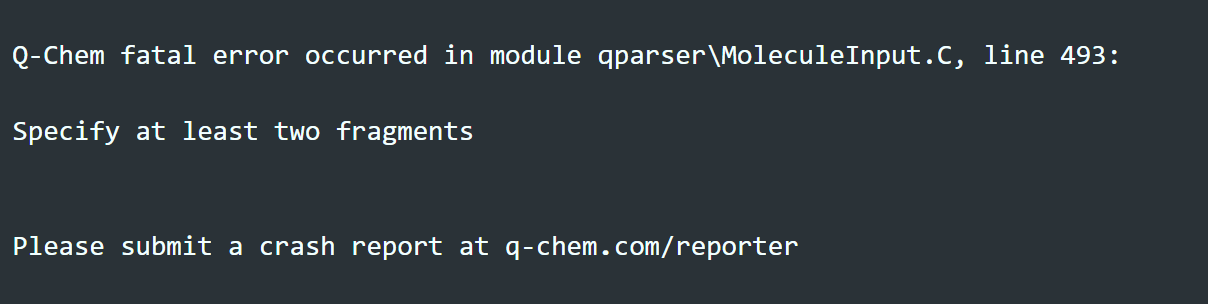
Dear all,
I’m using AIFDEM by Q-CHEM 6.0.2 windows version and want get dimer state like TT state or CT state.
I see the webinar 49,and usint the water dimer input 2, like
$rem
BASIS 6-31g*
EXCHANGE HF
CIS_N_ROOTS 1
CIS_SINGLETS TRUE
CIS_TRIPLETS TRUE
AIFDEM TRUE
AIFDEM_EMBED_RANGE 0
AIFDEM_NTOTHRESH 100
NTO_PAIRS 1
AIFDEM_SINGFIS TRUE
AIFDEM_CTSTATES TRUE
xpol true
$end
$molecule
0 1
– frgm 0
0 1
h -0.6305 -0.8758 0.2588
O -1.5614 -0.8255 0.5368
h -1.9991 -1.5191 0.0288
0 1 frgm 1
O 1.2821 -0.7524 -0.1108
H 1.5168 0.1326 -0.4203
H 1.7973 -0.8645 0.6993
$end
but I cant get the expand basis state
and the output file got error.
How can I fix it?
Thanks
Your $molecule input section is formatted incorrectly. Try
$molecule
0 1
--
0 1
h -0.6305 -0.8758 0.2588
O -1.5614 -0.8255 0.5368
h -1.9991 -1.5191 0.0288
--
0 1
O 1.2821 -0.7524 -0.1108
H 1.5168 0.1326 -0.4203
H 1.7973 -0.8645 0.6993
$end
Also, in recent versions of Q-Chem you will need to specify the choice of embedding charges since the XPol default (=CM5) is not implemented for AIFDEM. There will be a separate error message about this, you can try something like
$xpol
charges mulliken
$end
Dear jherbert,
Thank you, I use
$molecule
0 1
–
0 1
h -0.6305 -0.8758 0.2588
O -1.5614 -0.8255 0.5368
h -1.9991 -1.5191 0.0288
0 1
O 1.2821 -0.7524 -0.1108
H 1.5168 0.1326 -0.4203
H 1.7973 -0.8645 0.6993
$end
$rem
EXCHANGE HF
BASIS 6-31G
cis_n_roots 1
cis_singlets true
cis_triplets true
AIFDEM TRUE
AIFDEM_EMBED_RANGE 0
AIFDEM_NTOTHRESH 90
NTO_PAIRS 1
AIFDEM_SINGFIS TRUE
AIFDEM_CTSTATES TRUE
$end
When turn the xpol off i can run the input file.
Yes, because XPol defaults to CM5 charges which are not implemented for AIFDEM. The default is modifiable as indicated above, or you can simply turn off embedding charges as you have done.