We are trying to use the oniom method of Q-Chem. Specifically, we placed small water cluster in a carbon nanotube. We specify small water cluster as QM part using the wb97x level of theory and also specify nanotube as MM part using oplsaa force field. In particular, The carbon and hydrogen atom types in nanotubeuse use benzene model, as shown in the attached letter, while oxyen and hydrogen atom types in the small water cluster is based on TIP3P model. out file as follows:
**** Step 1: MM calculation on the entire system ****
OPLSAA force field loaded.
Determining angles, torsions, etc. from user defined input
imptor test 0: 5-6-5-6
imptor test 0: 5-6-5-6
imptor test 0: 6-5-5-6
imptor test 0: 6-6-5-5
imptor test 0: 6-5-5-6
imptor test 0: 6-6-5-5…
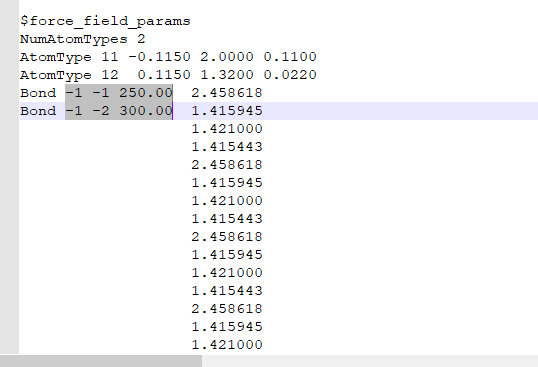
Therefore,we added force_field_params. However, how are these parameters set, especially force constant and epsilon?
