Hello!
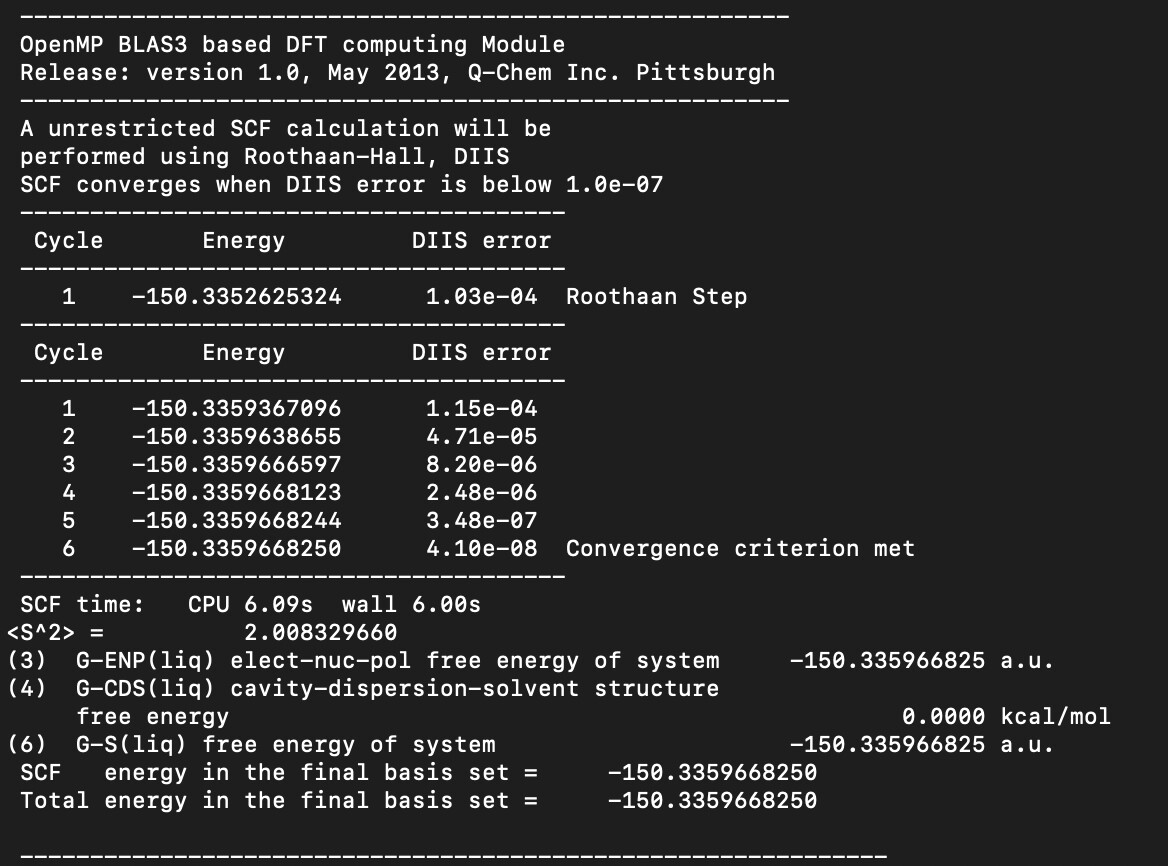
My calculations performed using SMD solvation model (QChem manual section 12.2.8.3 The SMD Model) are giving a cavity-dispersion-solvent structure free energy (G-CDS) of 0.0000 kcal/mol for O2 (and also with O and O3) in water with example output below. The resulting solvation free energy with a zero G-CDS differs from experimental values by ~2.5 kcal/mol.
The G-CDS and solvation free energies obtained when testing other molecules like methane or methanol match within 1 kcal/mol of the reference data reported the Minnesota group (who developed the model Minnesota Solvation Database (MNSOL) version 2012).
The solvent accessible surface areas are non-zero, so I do not expect a zero value for the G-CDS value (as defined in https://pubs.acs.org/doi/10.1021/jp810292n) unless the atomic surface tensions (defined therein; denoted sigma_k) are zero for O atoms.
Could you please advise why the G-CDS value is returning 0.0000 kcal/mol for these O calculations, and how I might obtain more accurate solvation energies?
I am using QChem 5.4.1
input parameters:
$molecule
0 3
O 0.5995474 0.1216532 0.0000000
O -0.7240777 -0.0241227 0.0000000
$end
$rem
BASIS = 6-311g++(2df,2p)
GUI = 2
JOB_TYPE = Optimization
METHOD = wB97M-V
SCF_CONVERGENCE = 7
THRESH = 12
Unrestricted = 1
Solvent_method = SMD
$end
output
Thank you for any suggestions that you can offer!
I’m not sure this is a bug. If you use
$smx
print 2
$end
you will get some additional info. I tried this for your O2 example and also for F2 and N2. Both O2 and N2 give G(CDS)=0.0 but for F2, I get (after turning on that additional printing):
Single-atom surface tensions:
------------------------------------------------
Element SASA Sigma Z Total
Z Ang**2 cal/Ang**2/mol kcal/mol
------------------------------------------------
F 59.589 38.180 2.275
Subtotal: 59.589 2.275
------------------------------------------------
In Table 4 of the paper that you reference, you will find 38.18 as the atomic surface tension parameter (sigma) for F in water. You will also notice that this parameter is omitted for both N2 and for O2, consistent with the fact that the code returns zero for the CDS term, for both molecules. If I do O2 in a different solvent, e.g.,
$smx
print 2
solvent thf
$end
then I do get a non-zero CDS term that comes from the “universal” solvation parameters.
Note that SMD is just a model, there is no reason that it should afford the experimental solvation energy. The mean accuracy of SMD for charge-neutral aqueous solutes is 0.9 kcal/mol (see Table 4 of https://doi.org/10.1002/wcms.1519), whereas I get 0.3 kcal/mol at the HF/6-31G* level. That might not be that far outside the distribution of errors (not sure what the variance is in the error statistics, I don’t easily find it in the SMD paper).
Thank you so much for sharing the print 2 option, pointing my attention to table 4 (and the absence of Sigma Z for O) so I can review more carefully, and sharing your insight about the expected errors and utility of the SMD model. I will consider more carefully whether this error is acceptable for my purposes.
Cheers! This was a great help.
No problem. It’s not obvious why it is that way, but at least based on cursory inspection it suggest that the code is consistent with the published model.