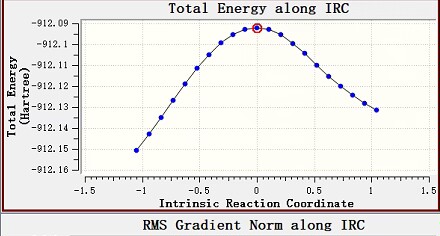
We have successfully upgraded to Q-Chem 6.0.2, but it does not seem to have solved our current problems. How do we get the mass-weighted coordinate on the one hand, or do we know how to calculate this coordinate from mass and coordinates? We have not found any papers on this and need your help urgently. Secondly, we find that the current output file contains only three step structures, are these three structures on the reaction path or are they the reactant and product structures inferred from the transition state? If it is the latter, that would be terrible as this is not the reactant and product structures we have entered. Our current input file is as follows:
$rem
JOBTYPE ts
BASIS aug-cc-pVTZ
METHOD pbe0
DFT_D = D3_BJ
MAX_SCF_CYCLES 25000000
GEOM_OPT_DMAX 50
GEOM_OPT_MAX_CYCLES 10000000000
GEOM_OPT_TOL_DISPLACEMENT=60
GEOM_OPT_TOL_GRADIENT 20
GEOM_OPT_TOL_ENERGY 20
SYMMETRY false
SYM_IGNORE true
$end
@@@
$molecule
read
$end
$rem
JOBTYPE freq
METHOD pbe0
DFT_D = D3_BJ
BASIS aug-cc-pVTZ
SCF_GUESS read
$end
@@@
$molecule
read
$end
$rem
JOBTYPE rpath
BASIS aug-cc-pVTZ
METHOD pbe0
DFT_D = D3_BJ
SCF_GUESS read
RPATH_MAX_CYCLES 250000000
$end