I have followed the Q-Chem menu to perform the IRC calculation.
However, there is a FileMan error in my calculation:
" Q-Chem fatal error occurred in module libmdc/newfileman.C, line 384:
FileMan error: End of file reached prematurely reading (38088) bytes in file FILE_NUCLEAR_HESSIAN
Path: /tmp/TS_cpp_irc/132.0
Please submit a crash report at q-chem.com/reporter "
Have you seen this problem and how to solve it?
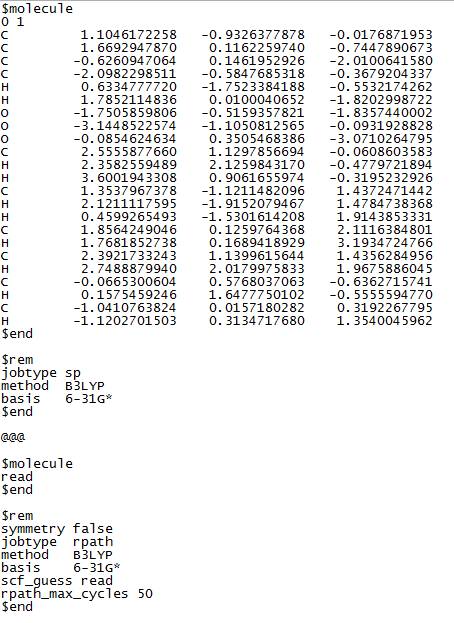
The problem is that rpath/IRC calculations require an initial (exact) Hessian, and Q-Chem is looking for the file containing the Hessian but can’t find it. From 9.6 Intrinsic Reaction Coordinate‣ Chapter 9 Exploring Potential Energy Surfaces: Searches for Critical Points and Molecular Dynamics ‣ Q-Chem 5.3 User’s Manual,
In order to use the IRC code, the transition state geometry and the exact Hessian must be available.
In reality, you can use any geometry and I believe any method that generates a Hessian and saves it to disk, but generally this means performing a frequency calculation beforehand, so replace jobtype sp in your first input with jobtype freq, if that indeed is a transition state.
1 Like
Thanks, I have recalculated the TS use large THRESH value, and then the irc calculation is ok.