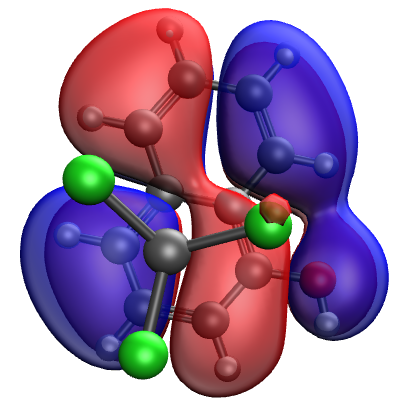
However, when switching to an EOM-CCSD calculation, in the lowest a few virtual orbitals I was not able to find an orbital that looks alike. The resulting excited state of a similar energy to the CT state obtained from TDDFT corresponds to a transition to
I was wondering if it is valid to use orbitals obtained from a DFT calculation (or other sources) to do EOM-CC calculations when the HF virtuals are of no good quality. And if it is, is EOM_CANONIZE = FALSE the only rem variable required to launch such a calculation?
Yes, it is possible to use molecular orbitals other than canonical Hartree-Fock orbitals. By default, such orbitals will be first canonicalized, which is controlled by CC_CANONIZE=TRUE/FALSE (TRUE is the default value). CC and EOM equations as implemented in Q-Chem deal correctly with the non-zero OV block of the Fock matrix, although one might observe poorer CCSD convergence than in the case of canonical orbitals.
To start with non-canonical orbitals, one must first obtain the orbitals and then run CC in another job in the input file:
Interesting example! There are several issues here.
To compare wave-functions and to assign characters of the states, you should not use raw orbitals but rather NTOs, as the latter deliver the essential picture of the excited state and are invariant wrt allowed orbital rotations (for TDDFT and EOM-CCSD, any unitary transformation within the occupied or virtual subspaces does not change the results). Using canonical KS or HF orbitals in the analysis can lead to erroneous conclusions about the state character. Fig. 8 in this paper http://iopenshell.usc.edu/pubs/pdf/jcp_perspective.pdf
illustrates this point.
If such CT state exists (and is not an artefact of the self-interaction error in DFT), EOM should find it regardless of the orbital choice. But some extra care and extra work might be needed – like increasing the number of guess vectors and roots, preconverging singles, etc (see the post in the HowTo section).
You can use EOM_USER_GUESS keyword to target an EOM state of the desired character, but this should be done with caution.
In some cases, it might be desirable to use other orbitals — for example, we often use KS orbitals in CC/EOM calculations of open-shell species to mitigate spin-contamination. This can be done by first running the job producing the desired orbitals followed (@@@) by a CC/EOM job with SCF_GUESS=read and SCF_MAX_CYCLES = 0 . I do not recommend to turn off EOM_CANONIZE, as it generally improves the convergence.
To add a bit to the above:
For the goals of effective Hamiltonians, I save the orbitals in which CC/EOM-CC is done, to an fchk file that Iqmol can visualize. This can be useful if the individual amplitudes are important. Just make sure that the you have a fresh Iqmol version.