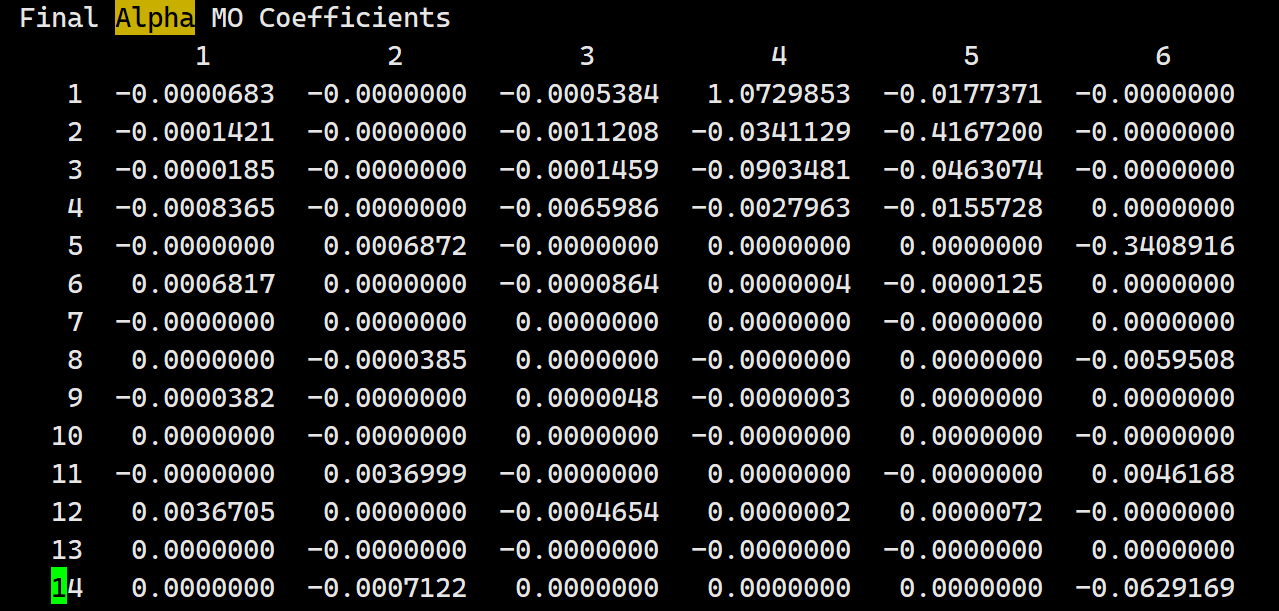
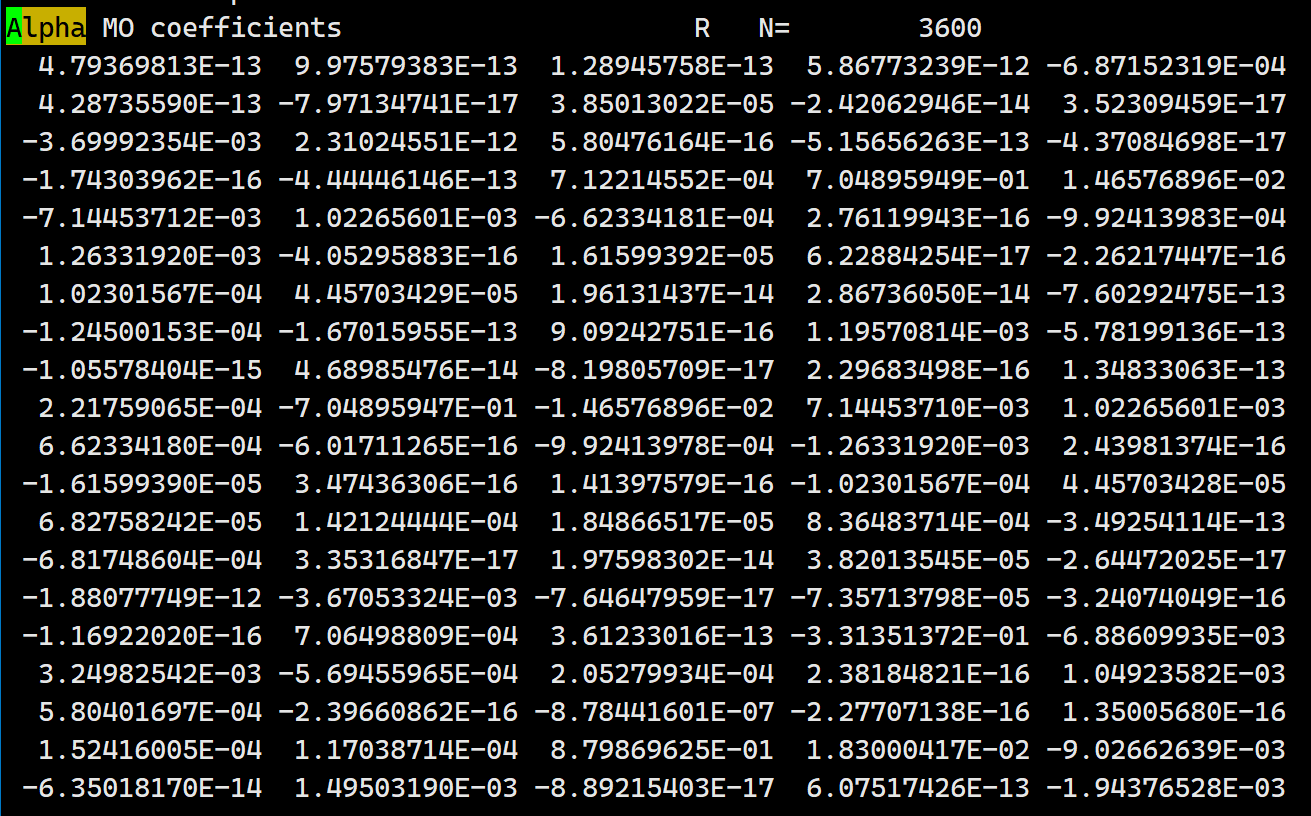
When using “scf_final_print 3” in the input file, the printed MO coefficients in the output file are completely different from those in the fchk or h5 file. What could be the reason for this?
Thank you very much for your reply. I believe it might be more convenient to find the answer from the source program. I have used other quantum chemistry software for the same calculation in order to verify the correct results, but unfortunately, the results obtained do not match the fchk and out files from QChem. I would like to ask if you can determine which results are correct (out or fchk)? This is important to me because I need to use this data for further calculations. I look forward to your reply, and once again, thank you very much!
It seems to be a problem with the compound job input (“@@@”). For the time being, please try running these as two separate jobs. Separate your input into job1.in and job2.in then do something like
(You can always run on multiple threads with -nt, not shown here.) In this case, everything seems to match; please confirm whether that’s true for you also. Note that you can use PRINT_ORBITALS = TRUE as an alternative to SCF_FINAL_PRINT = 3 to get MOs in a nicer output format. Note also that the MO coefficients in the .fchk file are transposed with respect to those in the Q-Chem output file.
Actually, I discovered this issue during a separate task. In order to facilitate communication, I merged the two tasks and reproduced the issue here. When I performed the separate task again just now, the second step still resulted in a mismatch between “out” and “fchk.” My Qchem software version is 6.1.1.
When using the keywords SCF_FINAL_PRINT 3 and PRINT_ORBITALS TRUE simultaneously, it seems that the orbitals printed by the two methods do not match. The orbitals printed by the PRINT_ORBITALS TRUE keyword match the fchk file, but the part printed by SCF_FINAL_PRINT 3 still does not match the previous two. Could this be due to the keywords? In addition, when testing with different systems, it was found that in some systems, using the SCF_FINAL_PRINT 3 keyword matches completely with the fchk file, while in others, it does not.
I would trust PRINT_ORBITALS, which is the standard way to get orbitals. Does this match the FChk in all cases?
In cases where SCF_FINAL_PRINT=3 gives something different, can you try setting GEN_SCFMAN = FALSE and do the same comparison? SCF_FINAL_PRINT options was added to GEN_SCFMAN (the new SCF code) relatively recently, maybe there’s a bug with that implementation.
Okay, I can reproduce this problem. Good news is that PRINT_ORBITALS matches the Fchk, whether I run this as a single compound input or as two sequential inputs. (Do you agree?) It is output from SCF_FINAL_PRINT=3 that fails to match, although strangely it does seem to match for certain orbitals but not the first ones. I suspect the latter is a bug. I will post a ticket on our developer site and see if someone else has some insight.
I would like to confirm that the orbitals coefficients printed by the keyword PRINT_ORBITALS always match with the fchk. However, the orbitals coefficients obtained with the keyword SCF_FINAL_PRINT=3 sometimes match with the fchk and sometimes do not.
Thank you very much for your attention.
When SCF_FINAL_PRINT=3 is used, is it possible that there is a hidden matrix diagonalization/iteration (e.g. 1 cycle of FC=SCE) between printing MO coefficients and creating .fchk file?
If such a matrix diagonalization/iteration exists, the MO coefficients of degenerate orbitals will probably be changed. Especially for the lowest core orbitals, they are usually degenerate. (For energetically degenerate orbitals, any unitary transformation does not change the orbital energy or the total electronic energy)
And if such a matrix diagonalization/iteration exists, there is one more problem: the DFT grid. The default grid may lead to near-degenerate orbitals for exactly degenerate ones. But if we add xc_grid 000099000590, the degenerate orbitals would just be degenerate. This numerical issue would affect the matrix diagonalization or reproduction of the problem.
The grid shouldn’t affect the agreement between the two printouts, but in light of your comment about an extra diagonalization, I increased SCF_CONVERGENCE to 8 in the 2nd job and now I get agreement between all three ways of accessing to coefficients. This does suggest that maybe the two printouts are out-of-sync by at least one Roothaan step.
Q-Chem staff looked into this further and it seems that it’s just swapping the order of two degenerate core orbitals. Depending on nuances like grid or convergence, the degeneracy might be split just enough for this not to happen. It’s a good question, made me think, but in the end it’s a feature (of quantum mechanics) not a bug.