I’m new to qchem and am trying to calculate the vibrational frequencies of the first excited state for CaCH3 with EOM-EA-CCSD and I couldn’t get rid of the negative frequency. I have also got the negative frequencies for CaOCH3 and CaNH2 while I could successfully calculate the FCFs of CaOCH2CH3. I guess it is probably because the geometry optimizetion converges to the saddle point. I have tried some commonly used trics such as tightening the convergence criteria and tweaking the geometry a little bit but that didn’t help.
Any suggestion is very helpful. Thank you very much in advance.
I attached a screenshot of my output for frequency job.
You probably need to break the symmetry of the initial guess structure. Despite the fact that you’ve turned symmetry off, it’s possible that the gradient has vanishing component along the symmetry-breaking mode. (Check the normal mode corresponding to the imaginary frequency and if I’m right, it will be one that breaks the symmetry.)
I tried a bunch of initial guess structures which break the symmetry, but all of them still give imaginary frequency. I moved atoms along thiese imaginary frequencies modes, but they all failed. Is there any suggenstion of how I should change the initial guess structure?
After I tried more, I was able to avoid negative frequencies, but when I computed FCFs of CaOCH3 with ezSpectrum, they are unrealistically non-diagonal. (The input for ezSpectra is below) I suspect it is because the geometries of ground and excited states are too different, and am wondering if the excited state geometry optimization converges to the correct geometry. Am I doing something wrong in the calculation of excited state? (input file for geometry optimization and freauency job for the excited state of CaOCH3 is also below)
optimization and frequency input:
$comment
optimization of first excited state (A2Pi), CaOCH3
O, C, H: aug-ccpvdz
Ca: aug-ccpwcvdz-pp + ECP28MDF
$end
$molecule
1 1
Ca
O 1 2.046460
C 2 1.407685 1 179.990605
H 3 1.108379 2 112.306270 1 -0.010000 0
H 3 1.108427 2 112.326352 1 122.051026 0
H 3 1.109427 2 110.326352 1 -118.051026 0
$end
$rem
BASIS = GEN
ECP = GEN
PURECART = 11
GUI = 2
JOB_TYPE = Optimization
METHOD = EOM-CCSD
EA_STATES = [2]
CC_STATE_TO_OPT = [1,2]
SCF_CONVERGENCE = 9
SCF_MAX_CYCLES = 1000
CC_CONVERGENCE = 9
EOM_DAVIDSON_CONVERGENCE = 9
GEOM_OPT_TOL_GRADIENT = 30
GEOM_OPT_TOL_DISPLACEMENT = 30
GEOM_OPT_TOL_ENERGY = 30
SYM_IGNORE = TRUE
$end
$basis
O 0
S 9 1.00
1.172000D+04 7.100000D-04
1.759000D+03 5.470000D-03
4.008000D+02 2.783700D-02
1.137000D+02 1.048000D-01
3.703000D+01 2.830620D-01
1.327000D+01 4.487190D-01
5.025000D+00 2.709520D-01
1.013000D+00 1.545800D-02
3.023000D-01 -2.585000D-03
S 9 1.00
1.172000D+04 -1.600000D-04
1.759000D+03 -1.263000D-03
4.008000D+02 -6.267000D-03
1.137000D+02 -2.571600D-02
3.703000D+01 -7.092400D-02
1.327000D+01 -1.654110D-01
5.025000D+00 -1.169550D-01
1.013000D+00 5.573680D-01
3.023000D-01 5.727590D-01
S 1 1.00
3.023000D-01 1.000000D+00
S 1 1.00
0.0789600 1.0000000
P 4 1.00
1.770000D+01 4.301800D-02
3.854000D+00 2.289130D-01
1.046000D+00 5.087280D-01
2.753000D-01 4.605310D-01
P 1 1.00
2.753000D-01 1.000000D+00
P 1 1.00
0.0685600 1.0000000
D 1 1.00
1.185000D+00 1.0000000
D 1 1.00
0.3320000 1.0000000
****
Ca 0
S 8 1.00
1.498270D+02 5.740000D-04
2.161640D+01 -1.781900D-02
1.351340D+01 8.618800D-02
4.438820D+00 -4.184470D-01
8.659740D-01 7.322470D-01
3.696370D-01 4.659510D-01
6.636900D-02 1.205000D-02
2.643200D-02 -3.605000D-03
S 8 1.00
1.498270D+02 -1.390000D-04
2.161640D+01 4.032000D-03
1.351340D+01 -2.143200D-02
4.438820D+00 1.142010D-01
8.659740D-01 -2.421620D-01
3.696370D-01 -3.152070D-01
6.636900D-02 6.660890D-01
2.643200D-02 5.010450D-01
S 1 1.00
1.023600D+00 1.000000D+00
S 1 1.00
3.696370D-01 1.000000D+00
S 1 1.00
2.643200D-02 1.000000D+00
S 1 1.00
1.050000D-02 1.000000D+00
P 7 1.00
3.443140D+01 2.171000D-03
7.588220D+00 -5.273000D-02
1.507550D+00 3.410420D-01
6.670350D-01 5.130630D-01
2.876280D-01 2.524570D-01
7.622300D-02 1.666700D-02
2.763300D-02 -2.381000D-03
P 7 1.00
3.443140D+01 -4.170000D-04
7.588220D+00 1.153900D-02
1.507550D+00 -8.253100D-02
6.670350D-01 -1.439400D-01
2.876280D-01 -6.530800D-02
7.622300D-02 5.495580D-01
2.763300D-02 5.588780D-01
P 1 1.00
1.081400D+00 1.000000D+00
P 1 1.00
2.763300D-02 1.000000D+00
P 1 1.00
1.000000D-02 1.000000D+00
D 5 1.00
9.089210D+00 3.782500D-02
2.390260D+00 1.552970D-01
7.068550D-01 3.099010D-01
1.937610D-01 4.499970D-01
5.052200D-02 4.766030D-01
D 1 1.00
1.394600D+00 1.000000D+00
D 1 1.00
5.052200D-02 1.000000D+00
D 1 1.00
2.020000D-02 1.000000D+00
****
H 0
S 4 1.00
1.301000D+01 1.968500D-02
1.962000D+00 1.379770D-01
4.446000D-01 4.781480D-01
1.220000D-01 5.012400D-01
S 1 1.00
1.220000D-01 1.000000D+00
S 1 1.00
0.0297400 1.0000000
P 1 1.00
7.270000D-01 1.0000000
P 1 1.00
0.1410000 1.0000000
****
C 0
S 9 1.00
6.665000D+03 6.920000D-04
1.000000D+03 5.329000D-03
2.280000D+02 2.707700D-02
6.471000D+01 1.017180D-01
2.106000D+01 2.747400D-01
7.495000D+00 4.485640D-01
2.797000D+00 2.850740D-01
5.215000D-01 1.520400D-02
1.596000D-01 -3.191000D-03
S 9 1.00
6.665000D+03 -1.460000D-04
1.000000D+03 -1.154000D-03
2.280000D+02 -5.725000D-03
6.471000D+01 -2.331200D-02
2.106000D+01 -6.395500D-02
7.495000D+00 -1.499810D-01
2.797000D+00 -1.272620D-01
5.215000D-01 5.445290D-01
1.596000D-01 5.804960D-01
S 1 1.00
1.596000D-01 1.000000D+00
S 1 1.00
0.0469000 1.0000000
P 4 1.00
9.439000D+00 3.810900D-02
2.002000D+00 2.094800D-01
5.456000D-01 5.085570D-01
1.517000D-01 4.688420D-01
P 1 1.00
1.517000D-01 1.000000D+00
P 1 1.00
0.0404100 1.0000000
D 1 1.00
5.500000D-01 1.0000000
D 1 1.00
0.1510000 1.0000000
****
$end
$ecp
CA 0
CA-ECP 4 10
g potential
1
2 1. 0.
s-g potential
2
2 10.556058402 138.832728745
2 5.293154907 16.754824599
p-g potential
4
2 12.714571445 27.661607611
2 12.878516057 55.297862576
2 4.147923622 4.210974307
2 3.841287457 7.627482586
d-g potential
4
2 4.703264136 -0.514324716
2 4.707975898 -0.765184799
2 13.696151450 -6.490621584
2 13.698165108 -9.735555558
f-g potential
2
2 14.820019137 -14.000148237
2 22.000803239 -61.616066332
****
$end
$molecule
READ
$end
$rem
BASIS = GEN
ECP = GEN
PURECART = 11
GUI = 2
JOB_TYPE = Frequency
METHOD = EOM-CCSD
EA_STATES = [2]
CC_STATE_TO_OPT = [1,2]
SYM_IGNORE = TRUE
SCF_CONVERGENCE = 8
SCF_MAX_CYCLES = 1000
$end
$basis
iO 0
S 9 1.00
1.172000D+04 7.100000D-04
1.759000D+03 5.470000D-03
4.008000D+02 2.783700D-02
1.137000D+02 1.048000D-01
3.703000D+01 2.830620D-01
1.327000D+01 4.487190D-01
5.025000D+00 2.709520D-01
1.013000D+00 1.545800D-02
3.023000D-01 -2.585000D-03
S 9 1.00
1.172000D+04 -1.600000D-04
1.759000D+03 -1.263000D-03
4.008000D+02 -6.267000D-03
1.137000D+02 -2.571600D-02
3.703000D+01 -7.092400D-02
1.327000D+01 -1.654110D-01
5.025000D+00 -1.169550D-01
1.013000D+00 5.573680D-01
3.023000D-01 5.727590D-01
S 1 1.00
3.023000D-01 1.000000D+00
S 1 1.00
0.0789600 1.0000000
P 4 1.00
1.770000D+01 4.301800D-02
3.854000D+00 2.289130D-01
1.046000D+00 5.087280D-01
2.753000D-01 4.605310D-01
P 1 1.00
2.753000D-01 1.000000D+00
P 1 1.00
0.0685600 1.0000000
D 1 1.00
1.185000D+00 1.0000000
D 1 1.00
0.3320000 1.0000000
****
Ca 0
S 8 1.00
1.498270D+02 5.740000D-04
2.161640D+01 -1.781900D-02
1.351340D+01 8.618800D-02
4.438820D+00 -4.184470D-01
8.659740D-01 7.322470D-01
3.696370D-01 4.659510D-01
6.636900D-02 1.205000D-02
2.643200D-02 -3.605000D-03
S 8 1.00
1.498270D+02 -1.390000D-04
2.161640D+01 4.032000D-03
1.351340D+01 -2.143200D-02
4.438820D+00 1.142010D-01
8.659740D-01 -2.421620D-01
3.696370D-01 -3.152070D-01
6.636900D-02 6.660890D-01
2.643200D-02 5.010450D-01
S 1 1.00
1.023600D+00 1.000000D+00
S 1 1.00
3.696370D-01 1.000000D+00
S 1 1.00
2.643200D-02 1.000000D+00
S 1 1.00
1.050000D-02 1.000000D+00
P 7 1.00
3.443140D+01 2.171000D-03
7.588220D+00 -5.273000D-02
1.507550D+00 3.410420D-01
6.670350D-01 5.130630D-01
2.876280D-01 2.524570D-01
7.622300D-02 1.666700D-02
2.763300D-02 -2.381000D-03
P 7 1.00
3.443140D+01 -4.170000D-04
7.588220D+00 1.153900D-02
1.507550D+00 -8.253100D-02
6.670350D-01 -1.439400D-01
2.876280D-01 -6.530800D-02
7.622300D-02 5.495580D-01
2.763300D-02 5.588780D-01
P 1 1.00
1.081400D+00 1.000000D+00
P 1 1.00
2.763300D-02 1.000000D+00
P 1 1.00
1.000000D-02 1.000000D+00
D 5 1.00
9.089210D+00 3.782500D-02
2.390260D+00 1.552970D-01
7.068550D-01 3.099010D-01
1.937610D-01 4.499970D-01
5.052200D-02 4.766030D-01
D 1 1.00
1.394600D+00 1.000000D+00
D 1 1.00
5.052200D-02 1.000000D+00
D 1 1.00
2.020000D-02 1.000000D+00
****
H 0
S 4 1.00
1.301000D+01 1.968500D-02
1.962000D+00 1.379770D-01
4.446000D-01 4.781480D-01
1.220000D-01 5.012400D-01
S 1 1.00
1.220000D-01 1.000000D+00
S 1 1.00
0.0297400 1.0000000
P 1 1.00
7.270000D-01 1.0000000
P 1 1.00
0.1410000 1.0000000
****
C 0
S 9 1.00
6.665000D+03 6.920000D-04
1.000000D+03 5.329000D-03
2.280000D+02 2.707700D-02
6.471000D+01 1.017180D-01
2.106000D+01 2.747400D-01
7.495000D+00 4.485640D-01
2.797000D+00 2.850740D-01
5.215000D-01 1.520400D-02
1.596000D-01 -3.191000D-03
S 9 1.00
6.665000D+03 -1.460000D-04
1.000000D+03 -1.154000D-03
2.280000D+02 -5.725000D-03
6.471000D+01 -2.331200D-02
2.106000D+01 -6.395500D-02
7.495000D+00 -1.499810D-01
2.797000D+00 -1.272620D-01
5.215000D-01 5.445290D-01
1.596000D-01 5.804960D-01
S 1 1.00
1.596000D-01 1.000000D+00
S 1 1.00
0.0469000 1.0000000
P 4 1.00
9.439000D+00 3.810900D-02
2.002000D+00 2.094800D-01
5.456000D-01 5.085570D-01
1.517000D-01 4.688420D-01
P 1 1.00
1.517000D-01 1.000000D+00
P 1 1.00
0.0404100 1.0000000
D 1 1.00
5.500000D-01 1.0000000
D 1 1.00
0.1510000 1.0000000
****
$end
$ecp
CA 0
CA-ECP 4 10
g potential
1
2 1. 0.
s-g potential
2
2 10.556058402 138.832728745
2 5.293154907 16.754824599
p-g potential
4
2 12.714571445 27.661607611
2 12.878516057 55.297862576
2 4.147923622 4.210974307
2 3.841287457 7.627482586
d-g potential
4
2 4.703264136 -0.514324716
2 4.707975898 -0.765184799
2 13.696151450 -6.490621584
2 13.698165108 -9.735555558
f-g potential
2
2 14.820019137 -14.000148237
2 22.000803239 -61.616066332
****
$end
Thank you very much for your information. Since I cannot get rid of this issue with Double zeta basis set, I am now trying with Triplet zeta basis sets. I have another question regarding memory so will create a separate discussion.
Since you are having difficulty with the cost of the triple-zeta calculation, I suggest perhaps not giving up on the double-zeta result. What is the nature of the imaginary mode?
I was playing around with your calculation (6-31G* basis and no ECP, so that it runs relatively quickly) and I noticed this message at the top of the frequency output:
!!.
!!Warning*.
!!Desired Analytical derivatives not available.
!!Finite difference job might take a long time.
!!.
!!********************************************.
2-order derivative to be evaluated numerically with 1-order analytical derivatives
EOM-IP/EA analytic gradients are available only for UHF references
Setting UNRESTRICTED=TRUE
It is very important to pay attention to what Q-Chem is telling you in the output file. Here, it indicates that it is setting UNRESTRICTED = TRUE, which I notice was not set in your optimization, which therefore ran with a RHF reference state. For my basis set, the UHF-based optimization takes a couple extra steps beyond the RHF-optimized structure, meaning that the geometries are not (quite) identical. This also means that according to the input that you posted above, you are running a UHF-based frequency job on top of an RHF-based geometry. Therefore the gradient is not guaranteed to be zero and you might wind up with imaginary frequencies.
Suggest that you redo the optimization with UNRESTRICTED=TRUE and the redo the frequency calculation at that geometry.
UPDATE: Setting UNRESTRICTED=TRUE for the optimization, I managed to obtain a structure with all real frequencies when BASIS=6-31G* (and no ECP). The rest of your input file was left the same.
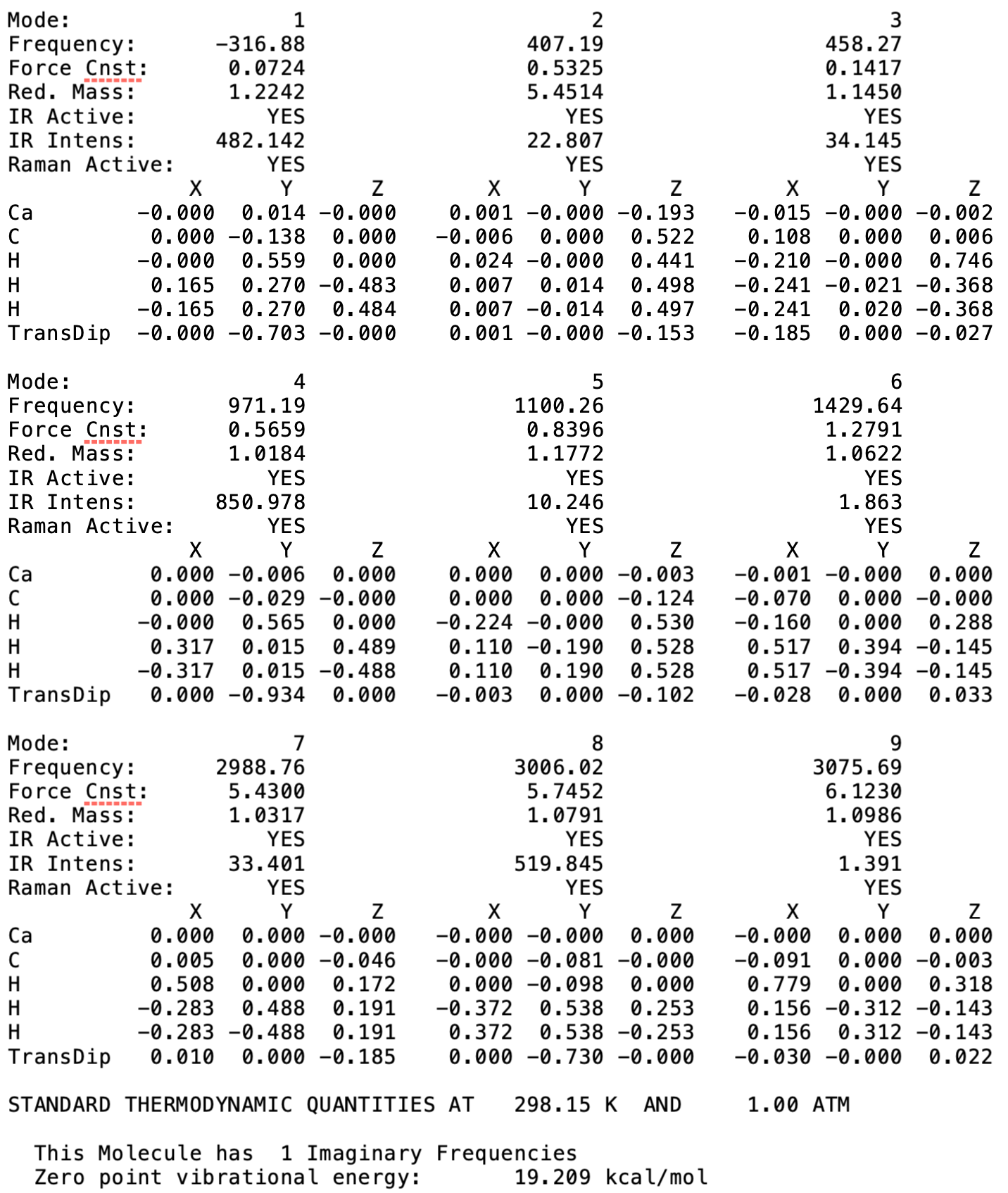
After I redid the optimization with UNRESTRICTED=TRUE and redid the frequency calculation at that geometry, I still got the negative frequency. (Please see the attached picture)
When I visualize this mode, this mode corresponds to the Ca-O-C bending motion that should be degenerate with mode 2 (Ca-O-C bending modes should be dooubly degenerate at frequency of ~ 135 cm^-1). I don’t know what this really means or suggests though…
Since you get real freqencies with 6-31G* basis, I am wondering if it is realted to the basis set for somehow. Do you think it is possible that smaller basis sets give imaginary frequency but larger basis sets don’t?
Could be related to the basis or could be related to the ECP, since I didn’t use one. The 6-31G* freq calculation takes about 20 min on 28 cores, here are the frequencies:
The 16cm^(-1) mode is Ca stretching, no surprise that is low-frequency, although I do not get the doubly-degenerate frequencies to which you refer. Was optimized w/o symmetry, here is the geometry:
1 Ca 0.0038360943 0.0087499152 0.0242886571
2 O 0.0000351213 -0.0035953537 2.0500448111
3 C -0.0025536583 -0.0119220887 3.4571941702
4 H 1.0238543493 0.0097771776 3.8550614060
5 H -0.5366651279 0.8638722622 3.8569982194
6 H -0.4952925515 -0.9161449595 3.8465967797
Thank you very much for sharing your calculation results. I think 16 cm^-1 is also too low according to the previous calculations and spectroscopies. The lowest freqeucny should be the Ca-O-C bending mode around 140 cm^-1 which should be doubly degenerate. So I don’t think the calculation isn’t going well with 6-31G* basis as well…
I just finished running the same calculation but with cc-pwCVTZ basis sets, and the screen shot below is what I got. There is a too low frequency mode which is simillar to what you had on your calculation. So it doesn’t seem to be related to ECP or diffuse function.
Is it possible that EOM-CCSD-EA has some trouble with symmetry breaking vibrational modes in C3v molecules? (I had no problems with linear molecules and was able to get the correct symmetry-breaking bending mode frequency in linear molecules like CaOH for example)
I wouldn’t have expected that basis to give quantitative results. What I was trying to do, and what I will now leave you to do, is systematically pare things down until you get to the root of the problem. Usually not best to jump into a new problem with an expensive level of theory before trying some cheaper things first, to get one’s bearings. For example, if I do a HF/6-31G* or a B3LYP+D3/6-311G** calculation, I get two degenerate modes of 150 and 137 cm^(-1), respectively. Notice that with the EOM-EA-CCSD you are getting finite-difference frequencies, with a stepsize that is controllable but whose default value may not have been chosen with very low frequencies in mind. I was trying to help you get this to run; now it’s time for you to do some experimenting with the models.