I’m very new to Q-chem and computational chemistry in general, so please keep this in mind if I don’t use the correct terminology.
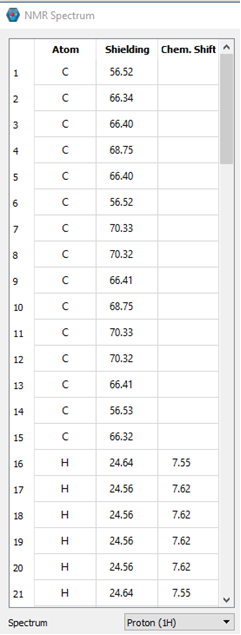
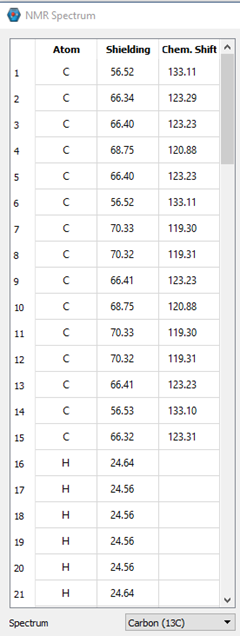
I calculated and NMR spectrum of a molecule of interest. IQmol displays the data fairly well. However, we’d like to “bin” similar atoms differently than what IQmol allows us to do. I’d also like to be able to zoom in on the graph, which I apparently cannot do in IQmol.
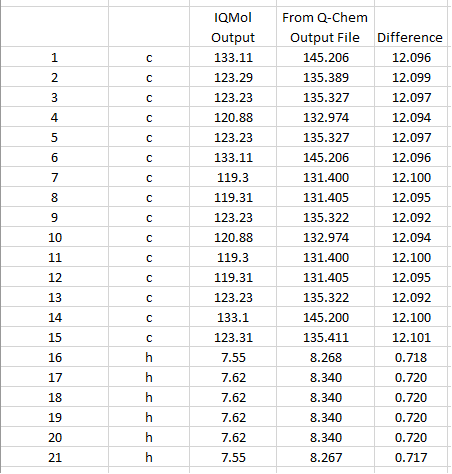
So my question is how do I get the chemical shifts out of the .out file? I see the table of relative chemical shifts for my Cs and Hs, but these values are not the same as what are displayed in IQmol. The values in IQmol are much closer to our measured values. The difference between IQmol’s display and the relative shifts is approximately 12.09 and 0.72 for the Cs and Hs, respectively.
Where do these correction values come from? Are these universal corrections, meaning the correction from relative shifts to what is displayed in IQmol will always be the same regardless of what method or basis set I use? Are these calculated somewhere and in the output file?
Hi Forrest,
I realize your molecule may be proprietary but could you provide some other toy example of a case where the numbers in the .out file don’t match what is plotted in IQmol? I don’t do NMR calculations so it’s hard for me to address this without a specific example to investigate.
Hi John,
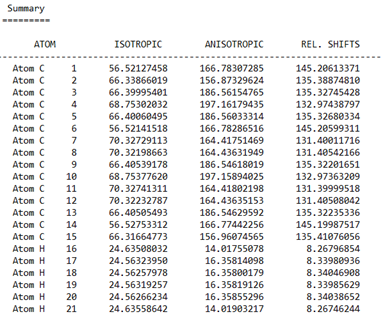
I can provide some information. There are 3 pictures below. The first shows some of the atoms from the output file and their relative shifts. The 2 pictures below that show what is displayed in IQmol. Based on our data and general knowledge of NMR, the values in IQmol are essentially the expected values. I’m sure there’s something simple I am missing or just don’t know about.
Note that the IQmol output and the Q-Chem output file differ by about 12.1 for carbon and 0.72 for H (see 4th picture at the bottom).
Thank you for taking a look!
From the Q-Chem output file:
From IQmol:
Direct comparison between IQmol and Q-Chem output file.
Thanks,
Forrest
Hi, maybe I can add a comment here.
The crucial thing to realise is the thing actually computed by Q-Chem is the absolute chemical shielding. This is under the column ISOTROPIC in qchem.out. And this is consistent, right?
The Chemical Shift is the difference between the absolute shielding of the molecule of interest vs the absolute shielding for the reference compound. So, it looks like Q-Chem and IQmol have different reference values.
The safest way is to compute the reference values yourself: Just do a computation on tetramethylsilane with exactly the same level that you are using for your molecule. Average over all the Cs / Hs and use this as the a reference value.
Obviously, the reference value depends on the computational method chosen. So, we are relying on tabulated values here and it seems that IQmol and Q-Chem use different tabulated values.
-Felix
1 Like
Hi Felix,
Thank you for the response.
I had wondered about the reference values, and yes, the reference values that are in my output file use a different method/basis set than what I used for my molecule of interest. So that is very likely the problem.
Quick additional question. Would you expect a chemical shift difference as large as 12 ppm between an HF/6-31G(d) calculation and a B3LYP/6-31G* calculation?
Thanks,
Forrest
Hi Felix,
I followed up with running TMS under the same level of theory (method and basis set) and sure enough, the calculations come out in much more realistic shifts.
Thank you so much for your help in figuring this out!
Forrest
ok! nice to hear that it works.
Regarding shifts with different methods, I am not quite sure. But it is reasonable to expect that Hartree-Fock would be off by quite a bit.
-Felix
Good to know.
Thanks again for your help. Have a great weekend.
Forrest
This is not my area of expertise but there is a comment in the Q-Chem manual (presumably written by someone who knows this stuff better) that Hartree-Fock chemical shifts are not of useful accuracy. What I do know is that chemical shifts, like other response properties, can be quite sensitive to basis set so 6-31G* is probably not what you want for production purposes. Jensen “polarization consistent” (pc-) basis sets were developed in part for NMR properties. See Table 7.9 here: Q-Chem 5.0 User’s Manual : Basis Set Symbolic Representation
Hi John,
Thank you for the insights. My goal with the NMR calculations at present was just to see if I could figure out how to do them as I expect to need to in the future. So I wanted to be sure I knew how to extract the correct chemical shifts, and I also wanted to be able to pull the data so that I can “bin” the data differently than the default in IQmol. I see how I can handle both, thanks to the help here!
Regarding choice of basis set, I was just following the basis set used in one of the lab handouts from the Q-Chem website (IR and NMR Spectra, Gilbert and Krylov). Page 4 lists the parameters to use, which I’m sure is good enough for the purpose of the lab handout…
“For structure (1), predict the number of peaks in the 1H and 13C NMR spectra. Compute
the NMR chemical shifts at the B3LYP/6-31G* level to confirm your predictions. (Click the
reset button in the QUI before setting up the job to remove the extra job sections.) Save
images for each of these spectra, you will need these to answer the questions.”