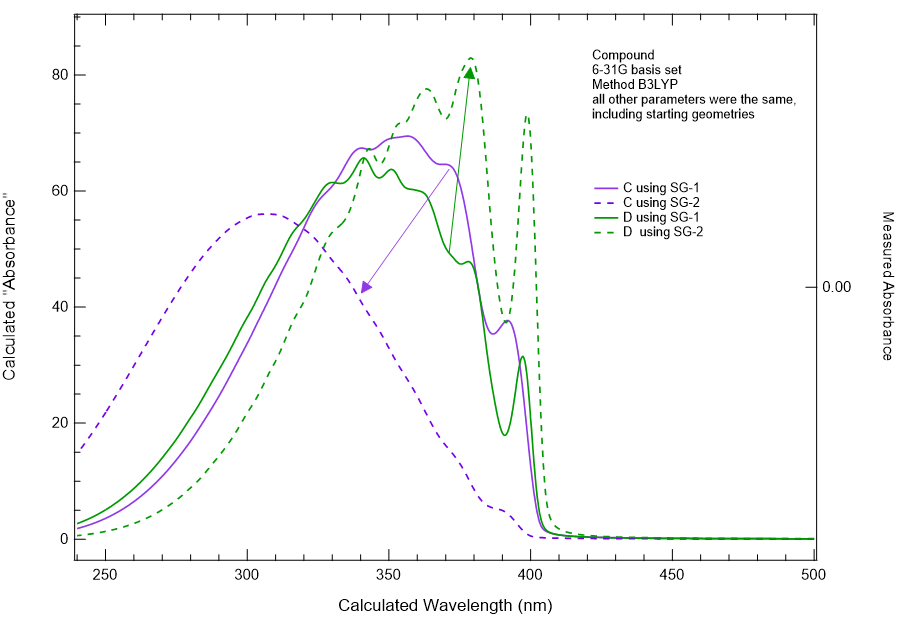
I’ve been using vibronic and changing the quadrature grid to calculate the UV-VIS spectrum of molecules of interest to us. I’ve found that sometimes when I go to the SG-2 grid from the SG-1 grid that I can get a huge improvement in the predicted spectrum. What I mean by huge improvement is that the spectrum shifts to about the right wavelength, and the shape of the spectrum narrows and looks more realistic. However, if I perform exactly the same calculation (SG-1 to SG-2) on the same molecule but different conformation (say a phenyl at +/- 35 degrees), I see dramatically different responses. In one case, my spectrum shifts bathochromically by 60 or more nm and structure appears such that the spectrum is starting to look like reality. However, with another conformation (yes, both conformations were minimized and look reasonable visually), I saw a hypsochromic shift of ~50 nm and a total loss of almost all structure.
I’m very surprised by this. I’m also very surprised by the dramatic differences in spectral shapes between conformers (think biphenyl going from +/- 35 degrees, though our compound is larger). The shape changes in the spectrum don’t make a lot of sense to me, chemically.
More than likely, there’s some other feature in Q-Chem that I’m missing, but I don’t know what that is. Any help would be greatly appreciated.
Forrest
This is a little surprising to me also, although in principle SG-2 is the higher-quality grid. What is the functional that you are using? The default grid is set sensibly for energies but occasionally for frequencies it needs to be tightened up.
Hi John,
As this was some of my first attempts at calculations at OSC, we were keeping our level of theory a bit lower.
The method was B3LYP.
The basis set was 6-31G.
And then the only difference was using SG-1 versus SG-2.
All other parameters were identical, including the starting geometries/coordinates.
B3LYP is an easy-to-integrate functional so the grid defaults to SG-1. That said, SG-1 is designed for fast energy calculations and it’s possible that frequencies, or the interplay of frequencies with intensities in a vibronic spectrum, is more demanding. On theoretical grounds I would therefore be more comfortable with SG-2, which is a higher-quality grid (more points per atom). We designed SG-2 to be the default (even for single-point energies) for harder-to-integrate functionals such as the B97 family.
Hi John,
We have several examples where moving to SG-2 or even SG-3 has provided UV-VIS spectra in vibronic that are much more structured and much closer to the experimental data, considering that at present we have not added solvent contributions. But then we have the issue in calculation C where to my eyes, theSG-2 calculation has ended up being much worse in regards to representing the experimental measurement. These spectra are of the same molecule, but of different conformations. The conformations are not at all unrealistic or even expected to be hugely different from one another in terms of energy level. So it is very bothersome that we see such a large change in the vibronic spectrum, shifting in the wrong direction.
I do find it odd that the C/SG-2 calculation seems to lose all the vibronic structure. Simply based on my own experience with TDDFT, I find it hard to believe that vertical excitation energies computed with B3LYP changed very much in going from SG-1 (default) to SG-2, although obviously you can check. That leaves the vibrational frequencies as the culprit and if I were you, I would take a look at how those change when the grid is changed.
Thank you, John. I will take a closer look.