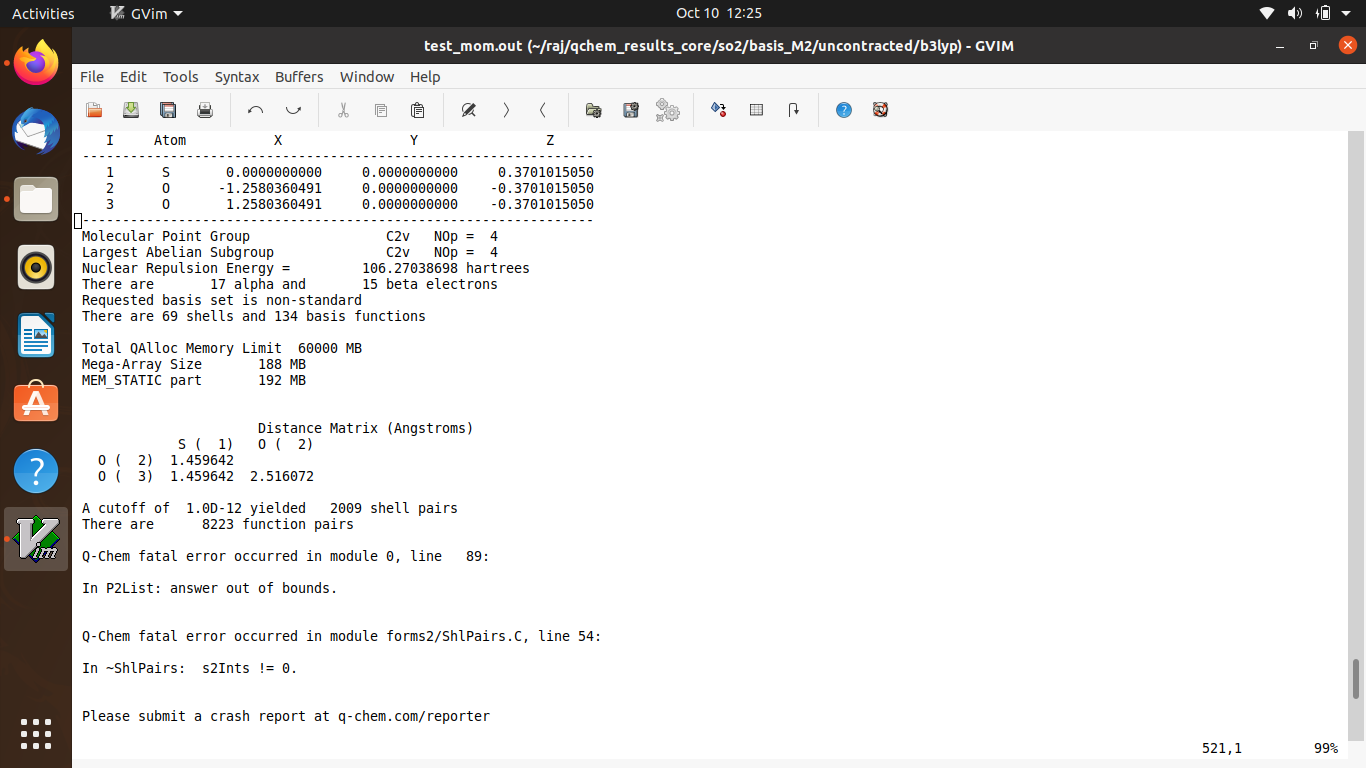
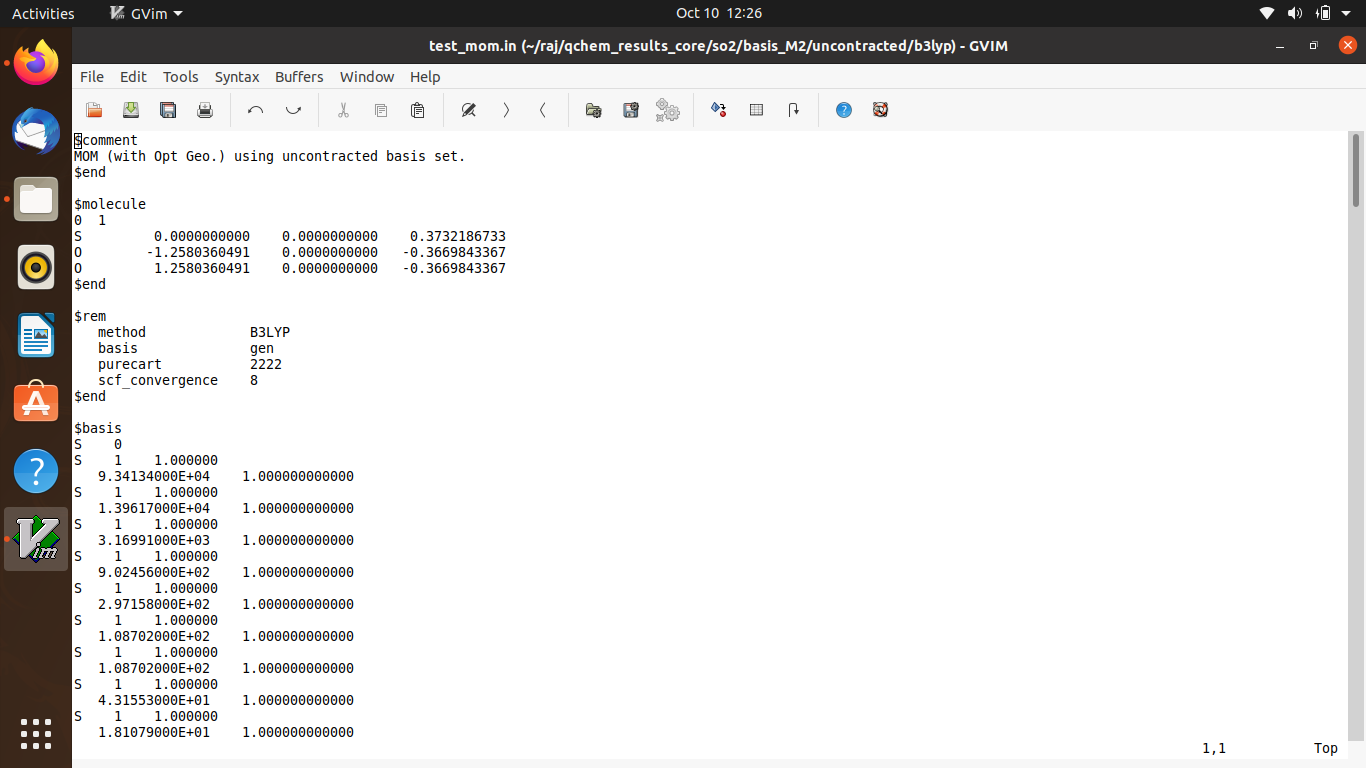
I was performing a DFT calculation by uncontracting the basis set for core excitation. But there is some error I believe which I am unable to fix.
"A cutoff of 1.0D-12 yielded… " is following at the end of the output file. I have also send a screen shot of the same.
Thank You
Can you please provide the input file?
Hi john, I have attached it in your email, since I cannot upload it here. Thanks for your reply.
Can you just copy/paste the text of the input file? I can’t diagnose this from a screen capture.
$comment
MOM (with Opt Geo.) using uncontracted basis set.
$end
$molecule
0 1
S 0.0000000000 0.0000000000 0.3732186733
O -1.2580360491 0.0000000000 -0.3669843367
O 1.2580360491 0.0000000000 -0.3669843367
$end
$rem
method B3LYP
basis gen
purecart 2222
scf_convergence 8
$end
$basis
S 0
S 1 1.000000
9.34134000E+04 1.000000000000
S 1 1.000000
1.39617000E+04 1.000000000000
S 1 1.000000
3.16991000E+03 1.000000000000
S 1 1.000000
9.02456000E+02 1.000000000000
S 1 1.000000
2.97158000E+02 1.000000000000
S 1 1.000000
1.08702000E+02 1.000000000000
S 1 1.000000
1.08702000E+02 1.000000000000
S 1 1.000000
4.31553000E+01 1.000000000000
S 1 1.000000
1.81079000E+01 1.000000000000
S 1 1.000000
5.56009000E+00 1.000000000000
S 1 1.000000
2.13183000E+00 1.000000000000
S 1 1.000000
4.20403000E-01 1.000000000000
S 1 1.000000
1.36045000E-01 1.000000000000
S 1 1.000000
4.05000000E-02 1.000000000000
S 1 1.000000
1.21987952E-02 1.000000000000
P 1 1.000000
4.95040000E+02 1.000000000000
P 1 1.000000
1.17221000E+02 1.000000000000
P 1 1.000000
3.77749000E+01 1.000000000000
P 1 1.000000
1.40584000E+01 1.000000000000
P 1 1.000000
5.56574000E+00 1.000000000000
P 1 1.000000
2.26297000E+00 1.000000000000
P 1 1.000000
8.07994000E-01 1.000000000000
P 1 1.000000
2.77460000E-01 1.000000000000
P 1 1.000000
7.71410000E-02 1.000000000000
P 1 1.000000
4.05000000E-02 1.000000000000
P 1 1.000000
1.21987952E-02 1.000000000000
D 1 1.000000
6.50000000E-01 1.000000000000
O 0
S 1 1.000000
8.58850000E+03 1.000000000000
S 1 1.000000
1.29723000E+03 1.000000000000
S 1 1.000000
2.99296000E+02 1.000000000000
S 1 1.000000
8.73771000E+01 1.000000000000
S 1 1.000000
2.56789000E+01 1.000000000000
S 1 1.000000
3.74004000E+00 1.000000000000
S 1 1.000000
4.21175000E+01 1.000000000000
S 1 1.000000
9.62837000E+00 1.000000000000
S 1 1.000000
2.85332000E+00 -1.000000000000
S 1 1.000000
9.05661000E-01 1.000000000000
S 1 1.000000
2.55611000E-01 1.000000000000
S 1 1.000000
8.45000000E-02 1.000000000000
S 1 1.000000
2.54518072E-02 1.000000000000
P 1 1.000000
4.21175000E+01 1.000000000000
P 1 1.000000
9.62837000E+00 1.000000000000
P 1 1.000000
2.85332000E+00 1.000000000000
P 1 1.000000
9.05661000E-01 1.000000000000
P 1 1.000000
2.55611000E-01 1.000000000000
P 1 1.000000
8.45000000E-02 1.000000000000
P 1 1.000000
2.54518072E-02 1.00000000E+00
D 1 1.000000
1.29200000E+00 1.00000000E+00
$end
@@@
$molecule
0 3
S 0.0000000000 0.0000000000 0.3732186733
O -1.2580360491 0.0000000000 -0.3669843367
O 1.2580360491 0.0000000000 -0.3669843367
$end
$rem
method B3LYP
basis gen
purecart 2222
mom_start 1
mom_print true
mom_method imom
scf_guess read
scf_convergence 8
unrestricted true
scf_final_print 2
max_scf_cycles 80
$end
$occupied
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
1 2 3 4 6 7 8 9 10 11 12 13 14 15 16
$end
$basis
S 0
S 1 1.000000
9.34134000E+04 1.000000000000
S 1 1.000000
1.39617000E+04 1.000000000000
S 1 1.000000
3.16991000E+03 1.000000000000
S 1 1.000000
9.02456000E+02 1.000000000000
S 1 1.000000
2.97158000E+02 1.000000000000
S 1 1.000000
1.08702000E+02 1.000000000000
S 1 1.000000
1.08702000E+02 1.000000000000
S 1 1.000000
4.31553000E+01 1.000000000000
S 1 1.000000
1.81079000E+01 1.000000000000
S 1 1.000000
5.56009000E+00 1.000000000000
S 1 1.000000
2.13183000E+00 1.000000000000
S 1 1.000000
4.20403000E-01 1.000000000000
S 1 1.000000
1.36045000E-01 1.000000000000
S 1 1.000000
4.05000000E-02 1.000000000000
S 1 1.000000
1.21987952E-02 1.000000000000
P 1 1.000000
4.95040000E+02 1.000000000000
P 1 1.000000
1.17221000E+02 1.000000000000
P 1 1.000000
3.77749000E+01 1.000000000000
P 1 1.000000
1.40584000E+01 1.000000000000
P 1 1.000000
5.56574000E+00 1.000000000000
P 1 1.000000
2.26297000E+00 1.000000000000
P 1 1.000000
8.07994000E-01 1.000000000000
P 1 1.000000
2.77460000E-01 1.000000000000
P 1 1.000000
7.71410000E-02 1.000000000000
P 1 1.000000
4.05000000E-02 1.000000000000
P 1 1.000000
1.21987952E-02 1.000000000000
D 1 1.000000
6.50000000E-01 1.000000000000
O 0
S 1 1.000000
8.58850000E+03 1.000000000000
S 1 1.000000
1.29723000E+03 1.000000000000
S 1 1.000000
2.99296000E+02 1.000000000000
S 1 1.000000
8.73771000E+01 1.000000000000
S 1 1.000000
2.56789000E+01 1.000000000000
S 1 1.000000
3.74004000E+00 1.000000000000
S 1 1.000000
4.21175000E+01 1.000000000000
S 1 1.000000
9.62837000E+00 1.000000000000
S 1 1.000000
2.85332000E+00 -1.000000000000
S 1 1.000000
9.05661000E-01 1.000000000000
S 1 1.000000
2.55611000E-01 1.000000000000
S 1 1.000000
8.45000000E-02 1.000000000000
S 1 1.000000
2.54518072E-02 1.000000000000
P 1 1.000000
4.21175000E+01 1.000000000000
P 1 1.000000
9.62837000E+00 1.000000000000
P 1 1.000000
2.85332000E+00 1.000000000000
P 1 1.000000
9.05661000E-01 1.000000000000
P 1 1.000000
2.55611000E-01 1.000000000000
P 1 1.000000
8.45000000E-02 1.000000000000
P 1 1.000000
2.54518072E-02 1.00000000E+00
D 1 1.000000
1.29200000E+00 1.00000000E+00
$end
Couple of things:
(1) Make sure there is a “****” (four asterisks) separating each atomic block of data in the $basis section. The software on this platform gobbles those up so I don’t see them, but I assume you have them there because Q-Chem generates a different error if not.
(2) While I do not understand the error that you are getting, it is associated with the symmetry code so my suggestion is to turn symmetry off (SYM_IGNORE=TRUE in $rem).
(3) I do not experience this error when running Q-Chem 6.0.1. (I do get SCF convergence failure but that’s a separate issue.)
Thanks John, sym_ignore=true is working…