Hello, everyone,I’m trying to calculate nmr shifting for a series of molecules. However, some of my tasks encounters fatal errors like
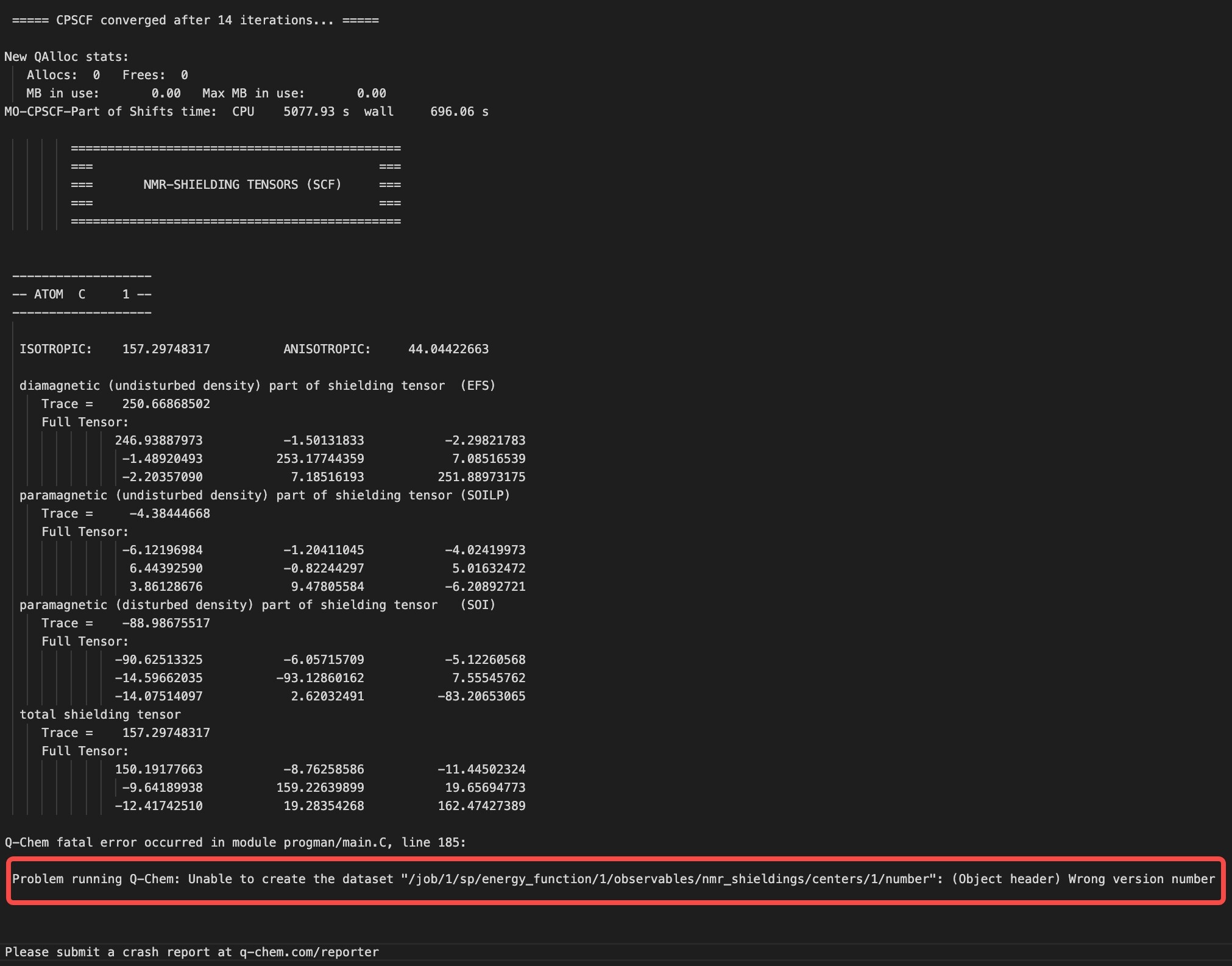
I re-run the tasks above and the errors can be reproduced, which seems to be related to my input file. However, I don’t know why the error comes from. So, does anyone know how to solve it.
my input is:
$molecule
0 1
C -1.81314969 -4.32687467 -2.71330386
C -2.39470084 -3.39055899 -1.65667714
C -2.89247130 -4.17737654 -0.44345598
C -3.48389673 -2.50108235 -2.25979013
O -1.24148068 -2.57187286 -1.28259188
C -1.34860255 -1.56776241 -0.39776034
O -2.34092893 -1.29071379 0.25394979
N -0.15709591 -0.91040853 -0.29727278
C -0.08516253 0.41072675 0.28617212
C -0.82936523 0.44879973 1.63810925
O -1.57718663 1.34639509 1.95375557
O -0.44631839 -0.55885880 2.40595699
C -1.03665059 -0.81094541 3.73064316
C -2.54916927 -0.98492689 3.59833680
C -0.37033845 -2.11928422 4.14618246
C -0.65752995 0.32661423 4.67852389
C -0.62934498 1.51435990 -0.67930806
O -1.66722133 1.09751124 -1.51169215
C -2.99386835 1.41586354 -1.08354786
C -3.22582287 2.90613091 -1.05705298
C -3.23042153 3.64180154 -2.25216933
C -3.39235791 5.02413950 -2.23034374
C -3.54585315 5.69632502 -1.01148111
Cl -3.74543068 7.42439434 -1.01227878
C -3.53077092 4.96963389 0.18846745
Cl -3.69511791 5.77193881 1.72356478
C -3.36583440 3.58088830 0.15903202
C 0.58351925 1.98781703 -1.51358374
N 0.78791349 1.14564031 -2.70558549
N -0.01803958 1.26541476 -3.62376682
N -0.67721412 1.30067481 -4.54902773
C 1.74948348 1.86021773 -0.54088512
C 2.42665096 0.51240744 -0.48480068
C 3.82983349 0.48834912 0.02814272
O 4.22744549 1.20159426 0.92329003
O 4.55651559 -0.41288241 -0.63796692
C 5.98142857 -0.63590968 -0.35221162
C 6.36294814 -1.72058584 -1.35644671
C 6.14779806 -1.13674402 1.08302317
C 6.76721009 0.64980475 -0.61250536
C 1.36191222 0.86949934 0.52603564
H -2.58840199 -5.01452090 -3.08289059
H -1.42260778 -3.74830626 -3.56353824
H -0.98994808 -4.91978848 -2.28788498
H -3.68353777 -4.87831785 -0.75206311
H -2.06871856 -4.76254082 -0.00595930
H -3.29680320 -3.50447891 0.32271043
H -4.28937137 -3.12958302 -2.66957459
H -3.06600001 -1.89425433 -3.07766966
H -3.90809326 -1.83126700 -1.50184456
H 0.52982701 -1.14377219 -1.00330138
H -3.03703651 -0.03764137 3.33988729
H -2.95805632 -1.34431793 4.55512902
H -2.77780027 -1.71576570 2.81043345
H 0.72401671 -2.00676532 4.15869199
H -0.70446099 -2.41455752 5.15209326
H -0.62842693 -2.92197662 3.43973330
H -1.01141421 0.09186120 5.69417329
H -1.10973591 1.27379209 4.35842221
H 0.43648411 0.44607420 4.71685984
H -0.92835838 2.36186852 -0.04412977
H -3.64617770 0.93384413 -1.82555415
H -3.19353165 0.96118159 -0.10407878
H -3.09945972 3.12503331 -3.20623394
H -3.40589656 5.60438986 -3.15420672
H -3.31459696 3.02888026 1.09854595
H 0.43830738 3.03481007 -1.82546504
H 2.34735863 2.73826762 -0.29178516
H 2.22330670 -0.18377691 -1.29804111
H 6.20011169 -1.36741108 -2.38550364
H 5.75409394 -2.62346257 -1.19961714
H 7.42388895 -1.98756303 -1.24064399
H 5.86700921 -0.36316556 1.80815849
H 7.19824470 -1.41770573 1.25403991
H 5.52383775 -2.02791915 1.25228371
H 7.84519351 0.44556926 -0.52338821
H 6.49495391 1.43302237 0.10546816
H 6.57037669 1.01578668 -1.63189216
H 1.71010861 1.00432433 1.55095516
$end
$rem
JOBTYPE FORCE
METHOD B3LYP
BASIS def2-SVP
DFT_D D3_BJ
SCF_CONVERGENCE 11
THRESH 14
MAX_SCF_CYCLES 50
RESP_CHARGES 1
HIRSHFELD true
SYMMETRY FALSE
SYM_IGNORE TRUE
MOPROP 1
MOPROP_PERTNUM 0
MOPROP_CONV_1ST 7
MOPROP_DIIS_DIM_SS 4
MOPROP_MAXITER_1ST 100
MOPROP_DIIS 5
MOPROP_DIIS_THRESH 1
MOPROP_DIIS_SAVE 0
WRITE_WFN 10004_neutral
$end