Hi there,
I would like to ask for advice on two problems I faced in the AIMD simulation under pressure.
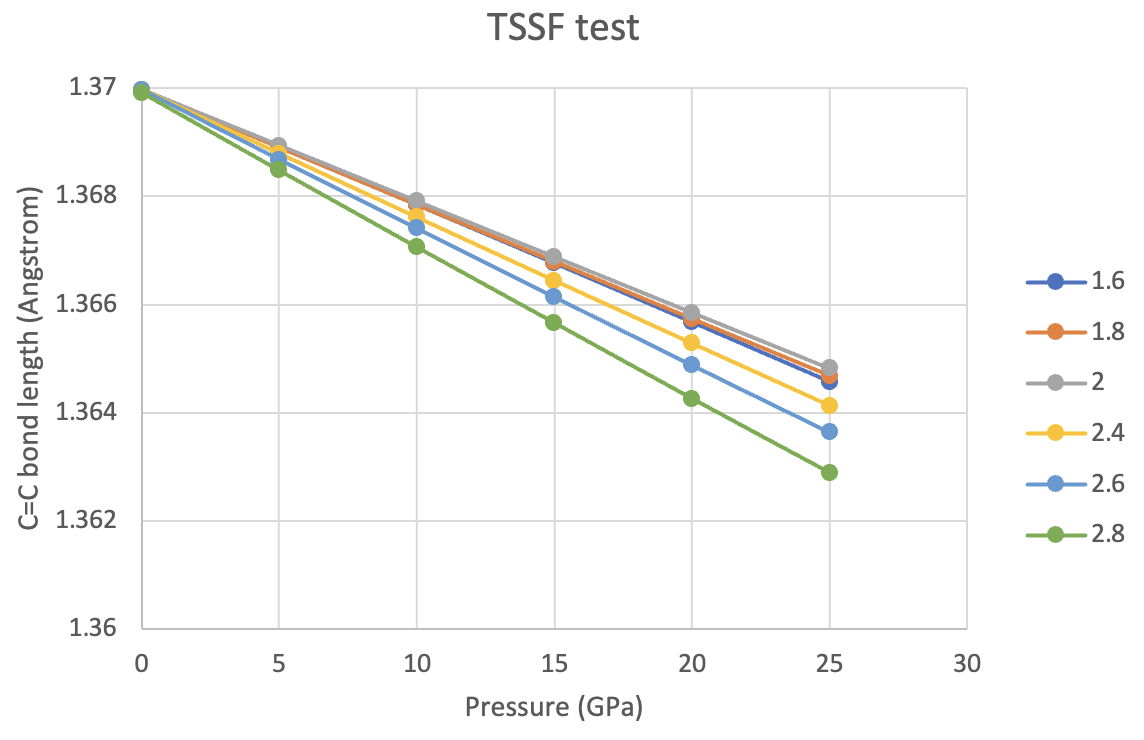
(1) When I did AIMD under pressure, what is the specific criteria for setting up the number of tessellation points and TSSF? I tried to set up a reaction with bigger molecule (anthracene) following the same protocol from topics " [Studying Molecules under External Pressure with the DISTORT module of Q-Chem]" and M. Scheurer et al., J. Chem. Theory Comput. 17, 583 (2021). The number of tessellation points seem to be converged while the results of TSSF test are still confusing for me.
(2) I also tried to rum the AIMD simulations with NTess = 302 and TSSF 1.8, but always got to wired structure or error like
Q-Chem fatal error occurred in module libgscf/gen_scfman/gen_scfman_main.C, line 245:
Error in gen_scfman
Is it because the calculation really depends on how close my initial guess should be to the equilibrium stage or just the van Der Waals cavity I set up is too small?
I also attached my opt input and one test graph.
Thanks a lot!
$molecule
0 1
C -3.5875270 -1.1458695 0.1437790
C -3.5941477 -0.3856334 -1.0646857
C -2.4100258 0.0569377 -1.6179501
C -1.1565851 -0.2183960 -0.9878142
C -2.3967634 -1.4452145 0.7735609
C -1.1498167 -0.9916074 0.2389851
C 0.0537752 0.3084112 -1.4741836
C 1.2668402 0.1252561 -0.7856765
C 0.0679271 -1.1991770 0.9114325
C 1.2745789 -0.6523648 0.4382583
C 2.4815975 0.7496710 -1.2086094
C 3.6400008 0.6216925 -0.4693334
C 3.6488235 -0.1486752 0.7325822
C 2.4975155 -0.7712565 1.1700100
H -4.5374099 -1.4835422 0.5759541
H -4.5490754 -0.1479605 -1.5491236
H -2.4078576 0.6584156 -2.5380547
H -2.3831576 -2.0143015 1.7140402
H 0.0438311 0.9191209 -2.3894159
H 0.0688824 -1.7629032 1.8563990
H 2.4627632 1.3554856 -2.1258858
H 4.5588702 1.1214261 -0.7992516
H 4.5746575 -0.2343645 1.3140457
H 2.4885823 -1.3483350 2.1054781
$end
$rem
symmetry off
sym_ignore true
jobtype opt
method b3lyp
basis 6-31G(d)
max_scf_cycles 500
gen_scfman true
distort true ! pressure is applied onto the molecule
scf_final_print 1 ! the energy contribution of the pressure is printed after SCF convergence
geom_opt_print 2
$end
$distort
model gostshyp ! the GOSTSHYP model is used
pressure 10000 !pressure = 40000 MPa = 40 GPa
npoints_heavy 302
npoints_hydrogen 302
scaling 1.6 ! non-default scaling factor is used
$end