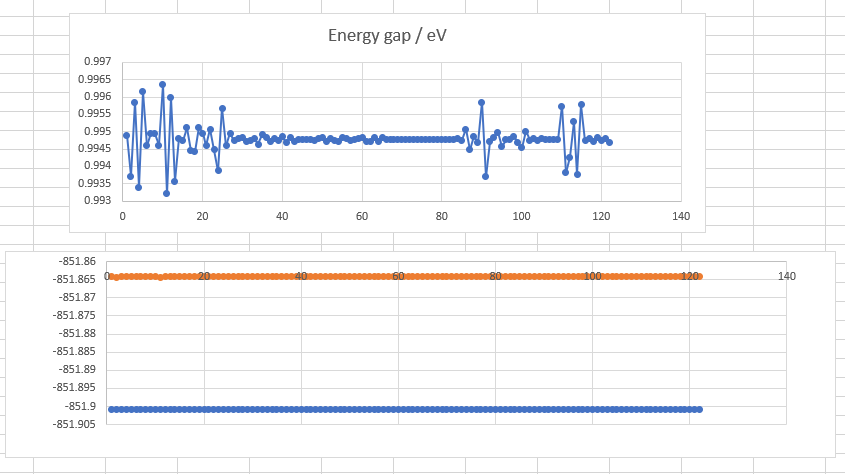
I tried to use SF-TDDFT to locate a TS of C=C bond rotation.
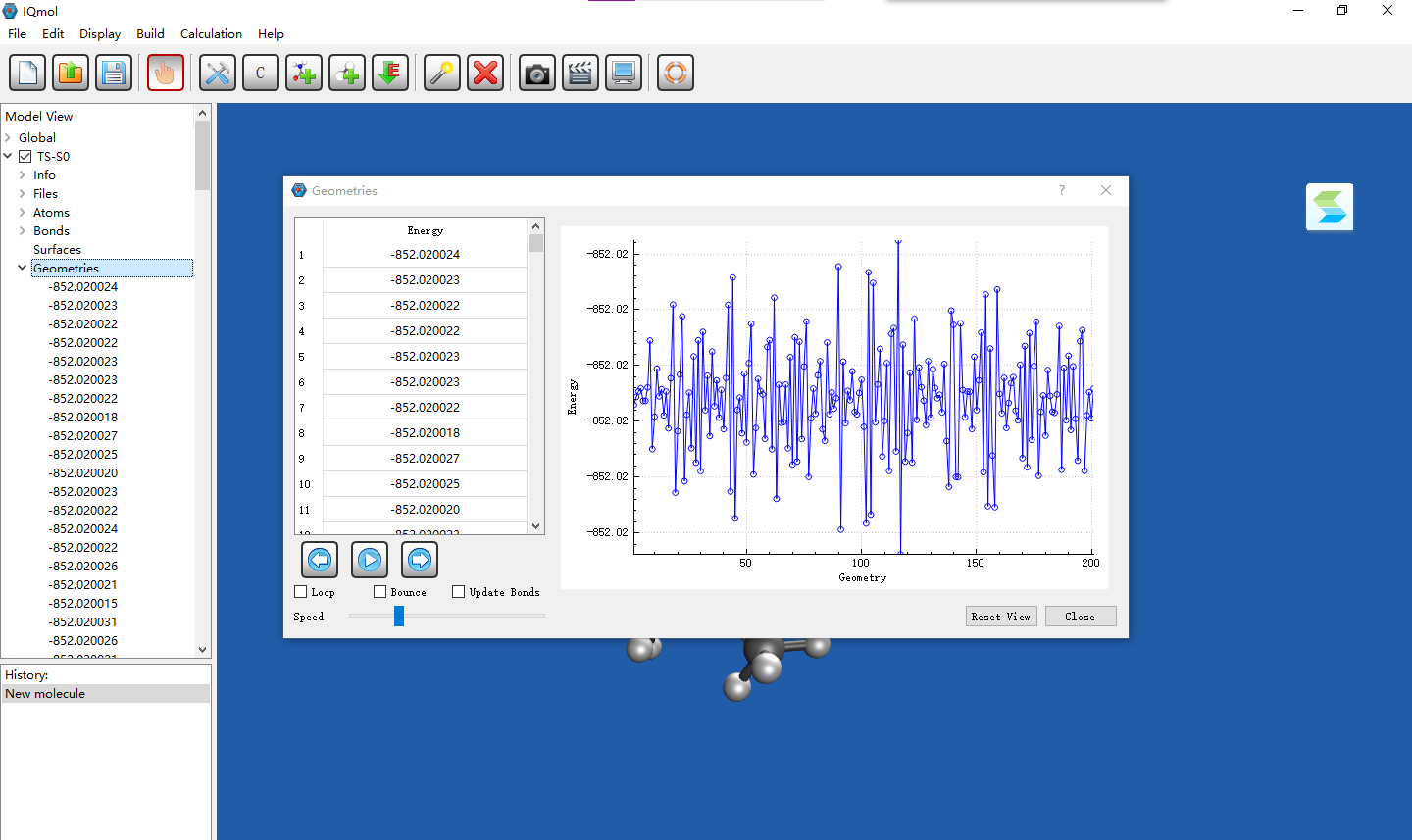
IQmol shows that there includes over 200 geometries (exactly the same). But the first job in input file is freq job which should only include one geometry. It also shows that total energy keeps oscillating.
Output file only shows “Running Job 1” and SCF is converged.
Thank you for your attention!
Below is my input:
$molecule
1 3
C -2.34670090 2.18774046 -0.13639317
C -3.84941067 2.15887725 0.16964645
H -4.03756893 1.84100530 1.20723421
H -4.27265805 3.16567783 0.04291284
H -4.39789125 1.48616254 -0.50611988
C -1.68447025 3.28315842 0.70527030
H -1.82535991 3.09694204 1.78109040
H -0.60570422 3.33969536 0.50352715
H -2.12719006 4.26085500 0.46543670
C -2.12986013 2.50266517 -1.62107132
H -1.06091177 2.59242920 -1.86738468
H -2.58068201 1.74967350 -2.28404096
H -2.59735229 3.46883652 -1.85992157
C -1.71542906 0.82363564 0.26387076
H -1.81977149 0.74631333 1.35759011
C -2.45531376 -0.36329497 -0.37300413
H -2.55924602 -0.22307705 -1.45757520
H -3.45127513 -0.49717068 0.06331145
N -0.29730476 0.70628576 -0.02363026
H -0.71741177 -1.37137932 -0.41306818
N -1.69134842 -1.63343169 -0.18231473
C -2.11950534 -2.70675255 -1.14113227
H -2.02022912 -2.27131413 -2.14286594
H -1.38564327 -3.51630335 -1.05704129
C -1.67474706 -2.06127449 1.25835964
H -1.16128728 -1.25798899 1.80141888
H -2.71913138 -2.08880338 1.59549774
C -0.96798011 -3.38232561 1.48514994
H -1.52870845 -4.23330746 1.07869969
H 0.04189379 -3.38060100 1.05133462
H -0.86755416 -3.53397920 2.56692360
C -3.52934872 -3.20190745 -0.89184506
H -4.27347339 -2.40170363 -0.99795954
H -3.75881317 -3.96972941 -1.64121851
H -3.63849643 -3.66249220 0.09914565
H -0.01738734 0.96931859 -0.96675210
C 0.67084141 0.91414632 0.96605985
C 2.01161356 1.36167801 0.55504807
C 3.01052758 0.40369586 0.16332932
C 4.23177347 0.79451007 -0.44650337
C 2.80304342 -0.98718365 0.36192721
C 5.17669869 -0.14874454 -0.83104243
H 4.43152531 1.84964041 -0.63435545
C 3.75508274 -1.92207433 -0.01872871
H 1.87875341 -1.31187033 0.84196618
C 4.95025022 -1.51265093 -0.62090132
H 6.10295846 0.18221470 -1.30404047
H 3.56746497 -2.98321709 0.15515709
H 5.69602612 -2.24858312 -0.92402646
H 0.29337490 1.07975312 1.97949923
C 2.25999072 2.83983395 0.43581221
H 1.62569566 3.39105271 1.14325432
H 3.30663687 3.10691387 0.63785551
H 2.01765477 3.21266191 -0.57698196
$end
$rem
jobtype freq
method bhhlyp
basis def2-svp
spin_flip true
unrestricted true
cis_n_roots 5
cis_triplets false
cis_state_deriv 1
solvent_method pcm
$end
$pcm
theory iefpcm
$end
$solvent
dielectric 35.9
opticaldielectric 1.803649
$end
@@@
$molecule
read
$end
$rem
jobtype ts
method bhhlyp
basis def2-svp
geom_opt_hessian read
geom_opt_max_cycles 128
spin_flip true
unrestricted true
cis_n_roots 5
cis_triplets false
cis_state_deriv 1
solvent_method pcm
$end
$pcm
theory iefpcm
$end
$solvent
dielectric 35.9
opticaldielectric 1.803649
$end
@@@
$molecule
read
$end
$rem
jobtype freq
scf_guess read
method bhhlyp
basis def2-svp
spin_flip true
unrestricted true
cis_n_roots 5
cis_triplets false
cis_state_deriv 1
solvent_method pcm
$end
$pcm
theory iefpcm
$end
$solvent
dielectric 35.9
opticaldielectric 1.803649
$end
@@@
$molecule
read
$end
$rem
jobtype rpath
scf_guess read
method bhhlyp
basis def2-svp
spin_flip true
unrestricted true
cis_n_roots 5
cis_triplets false
cis_state_deriv 1
rpath_max_cycles 50
solvent_method pcm
$end
$pcm
theory iefpcm
$end
$solvent
dielectric 35.9
opticaldielectric 1.803649
$end