X-ray lasers can trigger core ionization in a molecular system. The leaving electrons feel the electronic interaction in the molecular environment and thus carry the information of the electronic structure. Thanks to energy separation and locality of core ionizations, core photoelectron spectroscopy with X-ray has excellent element selectivity and spatial resolution, which makes it a powerful experimental tool in modern chemistry, physics, materials science and biology.
As correlation plays an important role in core ionization, high-level correlated methods such as coupled cluster (CC) theory are usually used to simulate core photoelectron spectroscopy (PES). CC and its equation-of-motion (EOM) variant are among the most accurate and versatile tools in studying core level spectroscopy. To consider ionizations, the EOM-IP-CC approach is adopted, in which the ground state is treated at the CC level and the ionized states are accessed by applying an ionization operator to the ground-state wavefunction. In order to reduce the computational cost, the CC calculations are done under the frozen core (fc) approximation, which means the ground state amplitudes and multipliers with occupied indices referring to a core orbital are set to zero and hence the core-core and core-valence correlation in the ground state is neglected. Since we only care about core ionizations here, the EOM states are represented in a restricted Fock space spanned by the many-electron determinants with at least one occupied core orbital index (the core-valence-separation approximation). The above sketches the main profile of the fc-CVS-EOM-CC approach.
Both ground states and excited state core PES can be studied by fc-CVS-EOM-CCSD. In ground state studies, the ionized states are obtained by fc-CVS-EOM-IP-CCSD, and the corresponding left- and right-Dyson orbitals are calculated, as the transformed Hamiltonian in the CC equations are not hermitian. The ionization intensities can be estimated using the product of norms of the left- and right-Dyson orbitals. The case of excited state core PES is different from that of ground states, since the final states of core ionization have double excitation character (2-hole-1-particle), which are not well described by at the level of EOM-CCSD. A scheme to circumvent this difficulty is to first obtain an excited triplet state with a valence hole through MOM, and then use this excited triplet state as a reference to calculate the core-ionized states and the corresponding left- and right-Dyson orbitals. Core ionizations from both alpha and beta channels show similar results.
In the following examples we show how to calculate the ground state PES of adenine and the excited state PES of uracil through fc-CVS-EOM-IP-CCSD.
Example. Simulating the C1s PES of the ground state of adenine. Geometry optimized at the MP2/cc-pVTZ level of theory.
Q-Chem input and comments (after “!”. Comments outside of the $rem section should be removed in order to run the qchem calculation.):
$molecule
0 1
N 1.663948 -1.127531 0.000000
N -0.643862 -1.804088 0.000000
N -0.636159 1.773662 0.000000
N -2.146840 0.119893 0.000000
N 2.353671 1.084678 0.000000
C 0.668787 -2.022720 0.000000
C -0.919981 -0.499328 0.000000
C 0.000000 0.546991 0.000000
C 1.356490 0.175959 0.000000
C -1.907009 1.474843 0.000000
H -3.039931 -0.341646 0.000000
H -2.712908 2.189994 0.000000
H 0.978346 -3.060960 0.000000
H 2.144452 2.065199 0.000000
H 3.305020 0.766647 0.000000
$end
$rem
print_general_basis = true
jobtype = sp
method = eom-ccsd
cvs_ip_states [5,0] ! calculate 5 ionized states in the first irrep, none for the second.
basis = general ! a general 6-311(2+,+)G** basis set is used (uncontracted core on C)
n_frozen_core = fc ! the frozen core approximation
cc_trans_prop = true ! calculate the transition properties of CC states
cc_do_dyson = true ! calculate Dyson orbitals
print_general_basis = true
molden_format = true ! output the orbitals in the MOLDEN format
$end
$basis
H 0
6-311(2+,+)G**
****
O 0
6-311(2+,+)G**
****
N 0
6-311(2+,+)G**
****
C 0
S 1 1.000000
4.56324000E+03 1.00000000E+00 ! uncontracted core on C
S 1 1.000000
6.82024000E+02 1.00000000E+00
S 1 1.000000
1.54973000E+02 1.00000000E+00
S 1 1.000000
4.44553000E+01 1.00000000E+00
S 1 1.000000
1.30290000E+01 1.00000000E+00
S 1 1.000000
1.82773000E+00 1.00000000E+00
SP 3 1.000000
2.09642000E+01 1.14660000E-01 4.02487000E-02
4.80331000E+00 9.19999000E-01 2.37594000E-01
1.45933000E+00 -3.03068000E-03 8.15854000E-01
SP 1 1.000000
4.83456000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
1.45585000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
4.38000000E-02 1.00000000E+00 1.00000000E+00
SP 1 1.000000
1.31927711E-02 1.00000000E+00 1.00000000E+00
D 1 1.000000
6.26000000E-01 1.00000000E+00
****
$end
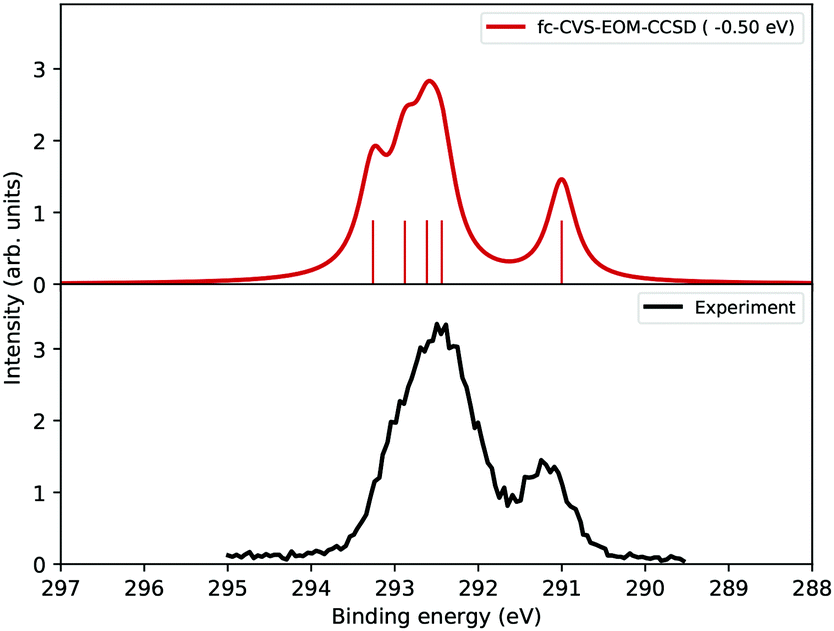
The simulation results are shown in the following figure:
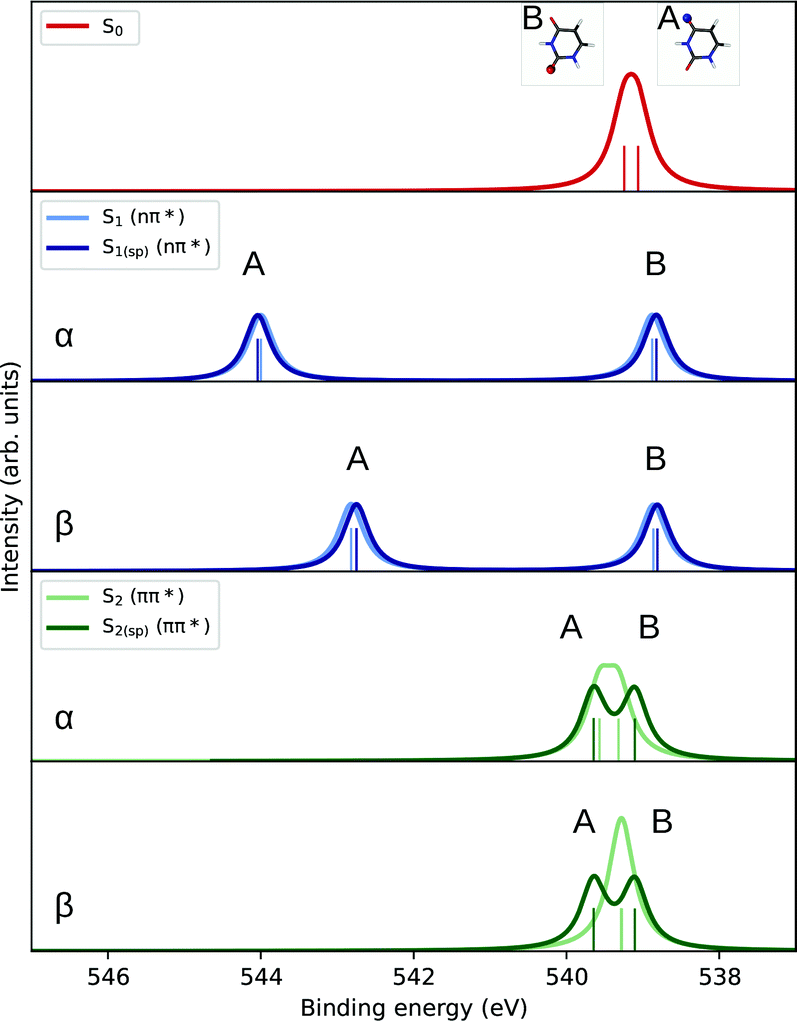
Example. Simulating the O1s PES of the excited state (S1) of uracil. Geometry optimized at the CCSD/aug-cc-pVDZ level of theory.
Q-Chem input and comments (after “!” in. Comments outside of the $rem section should be removed in order to run the qchem calculation.):
$comment
First step: a HF single point calculation of the neutral singlet state
$end
$molecule
0 1
C -0.0042112405 -1.7549930536 0.0000000000
H -2.0949460216 -1.4184138214 0.0000000000
H -0.1300971688 -2.8358204634 0.0000000000
H 0.0374086733 2.0243048927 0.0000000000
C -1.2210197500 0.4010705948 0.0000000000
O -2.2535781024 1.0567627444 0.0000000000
C 1.2399442445 0.2839850904 0.0000000000
O 2.3361887279 1.0691923037 0.0000000000
N 0.0349480223 1.0125648826 0.0000000000
N -1.1853832587 -0.9770870742 0.0000000000
C 1.2251302704 -1.1165150648 0.0000000000
H 2.1596039787 -1.6767971209 0.0000000000
$end
$rem
jobtype sp
basis general
method hf
n_frozen_core = 2 ! freeze 2 O1s electrons
$end
$basis
H 0
6-311(2+,+)G**
****
O 0
S 1 1.000000
8.58850000E+03 1.00000000E+00
S 1 1.000000
1.29723000E+03 1.00000000E+00
S 1 1.000000
2.99296000E+02 1.00000000E+00
S 1 1.000000
8.73771000E+01 1.00000000E+00
S 1 1.000000
2.56789000E+01 1.00000000E+00
S 1 1.000000
3.74004000E+00 1.00000000E+00
SP 3 1.000000
4.21175000E+01 1.13889000E-01 3.65114000E-02
9.62837000E+00 9.20811000E-01 2.37153000E-01
2.85332000E+00 -3.27447000E-03 8.19702000E-01
SP 1 1.000000
9.05661000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
2.55611000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
8.45000000E-02 1.00000000E+00 1.00000000E+00
SP 1 1.000000
2.54518072E-02 1.00000000E+00 1.00000000E+00
D 1 1.000000
1.29200000E+00 1.00000000E+00
****
N 0
S 1 1.000000
6.29348000E+03 1.00000000E+00
S 1 1.000000
9.49044000E+02 1.00000000E+00
S 1 1.000000
2.18776000E+02 1.00000000E+00
S 1 1.000000
6.36916000E+01 1.00000000E+00
S 1 1.000000
1.88282000E+01 1.00000000E+00
S 1 1.000000
2.72023000E+00 1.00000000E+00
SP 3 1.000000
3.06331000E+01 1.11906000E-01 3.83119000E-02
7.02614000E+00 9.21666000E-01 2.37403000E-01
2.11205000E+00 -2.56919000E-03 8.17592000E-01
SP 1 1.000000
6.84009000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
2.00878000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
6.39000000E-02 1.00000000E+00 1.00000000E+00
SP 1 1.000000
1.92469880E-02 1.00000000E+00 1.00000000E+00
D 1 1.000000
9.13000000E-01 1.00000000E+00
****
C 0
S 1 1.000000
4.56324000E+03 1.00000000E+00
S 1 1.000000
6.82024000E+02 1.00000000E+00
S 1 1.000000
1.54973000E+02 1.00000000E+00
S 1 1.000000
4.44553000E+01 1.00000000E+00
S 1 1.000000
1.30290000E+01 1.00000000E+00
S 1 1.000000
1.82773000E+00 1.00000000E+00
SP 3 1.000000
2.09642000E+01 1.14660000E-01 4.02487000E-02
4.80331000E+00 9.19999000E-01 2.37594000E-01
1.45933000E+00 -3.03068000E-03 8.15854000E-01
SP 1 1.000000
4.83456000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
1.45585000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
4.38000000E-02 1.00000000E+00 1.00000000E+00
SP 1 1.000000
1.31927711E-02 1.00000000E+00 1.00000000E+00
D 1 1.000000
6.26000000E-01 1.00000000E+00
****
$end
@@@
$comment
Second step: read in the MOs from the previous step, and obtain a neutral triplet excited state through MOM, then run CVS-EOM-IP-CCSD to calculate the ionized states
$end
$molecule
0 3 ! triplet configuration is considered as reference for core ionization
C -0.0042112405 -1.7549930536 0.0000000000
H -2.0949460216 -1.4184138214 0.0000000000
H -0.1300971688 -2.8358204634 0.0000000000
H 0.0374086733 2.0243048927 0.0000000000
C -1.2210197500 0.4010705948 0.0000000000
O -2.2535781024 1.0567627444 0.0000000000
C 1.2399442445 0.2839850904 0.0000000000
O 2.3361887279 1.0691923037 0.0000000000
N 0.0349480223 1.0125648826 0.0000000000
N -1.1853832587 -0.9770870742 0.0000000000
C 1.2251302704 -1.1165150648 0.0000000000
H 2.1596039787 -1.6767971209 0.0000000000
$end
$rem
job_type = sp
basis = general
print_general_basis = true
method = eom-ccsd
Scf_guess = read ! read in the MOs from the previous step
mom_start = 1 ! invoke MOM to obtain an excited state
cvs_ip_states = [4,0] ! calculate 4 ionized states in the first irrep, none for the second.
cvs_eom_shift = 19500 ! specifies energy shift in CVS-EOM calculations.
n_frozen_core = 2 ! freeze 2 O1s electrons
cc_trans_prop = true ! calculate the transition properties of CC states
cc_do_dyson = true ! calculate Dyson orbitals
molden_format = true ! output the orbitals in the MOLDEN format
$end
$basis
H 0
6-311(2+,+)G**
****
O 0
S 1 1.000000
8.58850000E+03 1.00000000E+00
S 1 1.000000
1.29723000E+03 1.00000000E+00
S 1 1.000000
2.99296000E+02 1.00000000E+00
S 1 1.000000
8.73771000E+01 1.00000000E+00
S 1 1.000000
2.56789000E+01 1.00000000E+00
S 1 1.000000
3.74004000E+00 1.00000000E+00
SP 3 1.000000
4.21175000E+01 1.13889000E-01 3.65114000E-02
9.62837000E+00 9.20811000E-01 2.37153000E-01
2.85332000E+00 -3.27447000E-03 8.19702000E-01
SP 1 1.000000
9.05661000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
2.55611000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
8.45000000E-02 1.00000000E+00 1.00000000E+00
SP 1 1.000000
2.54518072E-02 1.00000000E+00 1.00000000E+00
D 1 1.000000
1.29200000E+00 1.00000000E+00
****
N 0
S 1 1.000000
6.29348000E+03 1.00000000E+00
S 1 1.000000
9.49044000E+02 1.00000000E+00
S 1 1.000000
2.18776000E+02 1.00000000E+00
S 1 1.000000
6.36916000E+01 1.00000000E+00
S 1 1.000000
1.88282000E+01 1.00000000E+00
S 1 1.000000
2.72023000E+00 1.00000000E+00
SP 3 1.000000
3.06331000E+01 1.11906000E-01 3.83119000E-02
7.02614000E+00 9.21666000E-01 2.37403000E-01
2.11205000E+00 -2.56919000E-03 8.17592000E-01
SP 1 1.000000
6.84009000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
2.00878000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
6.39000000E-02 1.00000000E+00 1.00000000E+00
SP 1 1.000000
1.92469880E-02 1.00000000E+00 1.00000000E+00
D 1 1.000000
9.13000000E-01 1.00000000E+00
****
C 0
S 1 1.000000
4.56324000E+03 1.00000000E+00
S 1 1.000000
6.82024000E+02 1.00000000E+00
S 1 1.000000
1.54973000E+02 1.00000000E+00
S 1 1.000000
4.44553000E+01 1.00000000E+00
S 1 1.000000
1.30290000E+01 1.00000000E+00
S 1 1.000000
1.82773000E+00 1.00000000E+00
SP 3 1.000000
2.09642000E+01 1.14660000E-01 4.02487000E-02
4.80331000E+00 9.19999000E-01 2.37594000E-01
1.45933000E+00 -3.03068000E-03 8.15854000E-01
SP 1 1.000000
4.83456000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
1.45585000E-01 1.00000000E+00 1.00000000E+00
SP 1 1.000000
4.38000000E-02 1.00000000E+00 1.00000000E+00
SP 1 1.000000
1.31927711E-02 1.00000000E+00 1.00000000E+00
D 1 1.000000
6.26000000E-01 1.00000000E+00
****
$end
$occupied ! specify the orbital occupation
1:29 41 ! alpha orbitals no. 1-29 and 41 are occupied, which is specific to state S1
1:26 28 29 ! beta orbital no. 1-29 are occupied except no. 27, which is specific to state S1
$end
The simulation results are shown in the following figure: