Hello. I’m proceeding the geometry optimization calculations at the level of M06-2X-D/6-31G* considering empirical grimme dispersion. However, at the second optimization cycle, the molecule looks strange like below. The original connectivity between the atoms are broken and they are arranged disorderly. After this step, the molecule recoverd its structure similar to the initial one but I’m not sure whether there are problems or not. Can you diagnose this unusual situation?
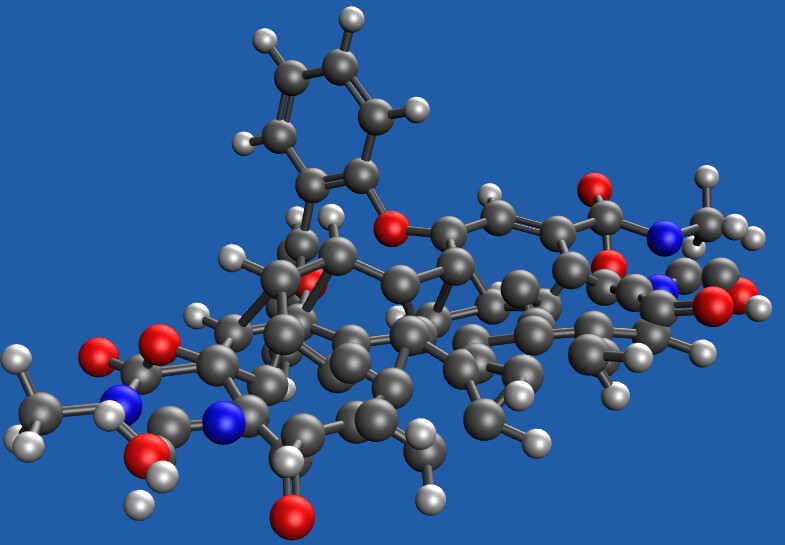
How reasonable is the starting geometry?
Could you please paste the starting coordinates so I could visualize it myself and rotate a bit to see how far the fragments are from each other?
Has the SCF cycle converged without problems for the first geometry iterations?
Some time ago I noticed that “D” correction for aromatic compounds tend to push condensed rings too close to each other. Probably, this happens here as well. I would recommend to try optimization without the dispersion correction and see what happens.
- Here are the xyz coordinates.
C -3.3881200475 2.0034744255 -0.7888631903
C -2.0370803351 1.6871845705 -0.9857387377
C -1.6040678282 0.4047976374 -1.3534294990
C -2.5987752427 -0.6331635314 -1.3574473861
C -3.9733508818 -0.3076938801 -1.1438736536
C -4.3537646062 1.0292753297 -0.8836878994
C -2.2392687223 -2.0120284856 -1.4971084390
C -3.2495421543 -2.9760210614 -1.4768632268
C -4.5941481584 -2.6397199463 -1.3007102310
C -4.9617252652 -1.3189606910 -1.1158783794
C -6.3839026913 -0.9974321548 -0.8393108711
N -6.6989591913 0.3492952124 -0.6346615232
C -5.7657713823 1.3896131818 -0.6098659821
C -0.2052709982 0.0487368015 -1.6396231076
C 0.1572188986 -1.3388357965 -1.6704424402
C -0.8222235990 -2.3762211147 -1.5908662929
C 1.5354766901 -1.6965076829 -1.7816531373
C 1.9232602396 -3.0555425017 -1.7350102359
C 0.9593684204 -4.0434199760 -1.6362856107
C -0.3953966778 -3.7053981954 -1.5835189983
C 0.7935679347 0.9922471213 -1.8912538242
C 2.1405457720 0.6284167759 -2.0226609311
C 2.5272736639 -0.6951647042 -1.9232908408
C 3.9663370428 -1.0445259679 -1.9624854378
N 4.2821622932 -2.4093401553 -1.9639269080
C 3.3533731723 -3.4384461163 -1.7642044772
O -7.2438802754 -1.8695611183 -0.7879459333
O -6.1175681973 2.5373378255 -0.3773670211
O 4.8486662900 -0.1922987214 -1.9833864026
O 3.7269535176 -4.5965685325 -1.6238067851
O -1.1357124418 2.6502949557 -0.6028233323
C 5.6954621461 -2.8024431757 -2.0495792625
C -8.0817958351 0.7177345685 -0.3097422477
H -3.6662841555 3.0147702352 -0.5128287634
H -3.0063355062 -4.0259894748 -1.5746615585
H -5.3580931880 -3.4083536235 -1.2691583239
H 1.2785149259 -5.0797792820 -1.6000374456
H -1.1139075121 -4.5110186612 -1.5026536641
H 0.5524907602 2.0389490678 -1.9617787377
H 2.9011943769 1.3875819741 -2.1682483514
H 5.7618696142 -3.7135001187 -2.6462565678
H 6.2471237131 -1.9853097878 -2.5131332617
H 6.1049717722 -2.9937170414 -1.0543774741
H -8.1332960738 1.0440586857 0.7337642153
H -8.7117020347 -0.1561370430 -0.4673808073
H -8.3894226862 1.5477553237 -0.9499581470
C -1.3527495533 3.8962393160 -2.6603119264
C -1.0897139625 5.0556465297 -3.3885889287
C -0.3920212901 6.1068153492 -2.7933476412
C 0.0319491762 5.9878651388 -1.4705099044
C -0.2183844405 4.8375662561 -0.7026431942
C -0.9266366092 3.7947293964 -1.3340844996
H -1.8926447566 3.0678996105 -3.1078221001
H 0.5871113772 6.7977819210 -1.0063396629
H -0.1715721036 7.0100851625 -3.3542695087
H -1.4243926207 5.1276684949 -4.4195449971
C 1.3514205511 3.8845796480 2.6736805083
C 0.9244058428 3.7901491044 1.3471808960
C 0.2087052991 4.8332504528 0.7244289719
C -0.0488530600 5.9757395425 1.5013999349
C 0.3754881891 6.0873852085 2.8247595584
C 1.0812040535 5.0365779425 3.4110672531
H -0.6101493461 6.7853072862 1.0440983202
H 0.1491843283 6.9846775793 3.3928898317
H 1.4165725906 5.1029705566 4.4421816448
H 1.8974538313 3.0566356010 3.1144031365
C -2.1380718121 0.6268329123 2.0213663989
C -0.7909672939 0.9907275180 1.8911365733
C 0.2078802374 0.0477669237 1.6377479387
C -0.1549022403 -1.3398416786 1.6657169361
C -1.5332271997 -1.6976028233 1.7759752482
C -2.5249932044 -0.6964477783 1.9191896812
C 0.8242980567 -2.3772321524 1.5841992040
C 0.3973690094 -3.7063557240 1.5745908580
C -0.9574024575 -4.0443517508 1.6266808585
C -1.9211750523 -3.0565074561 1.7268534260
C -3.3513322625 -3.4393429520 1.7550564837
N -4.2801051421 -2.4105014688 1.9560757423
C -3.9641389416 -1.0457088095 1.9570928744
C 1.6067424585 0.4043585592 1.3521107252
C 2.6011551815 -0.6340730072 1.3534984872
C 2.2413118465 -2.0131172541 1.4907888355
C 3.9758209200 -0.3089726343 1.1396655505
C 4.9637878605 -1.3206149073 1.1093732977
C 4.5957962715 -2.6415173614 1.2921238089
C 3.2511324438 -2.9774939028 1.4682547670
C 2.0403682723 1.6873211985 0.9867977010
C 3.3914618946 2.0029346711 0.7893115013
C 4.3567654167 1.0281445930 0.8814203153
C 5.7687178902 1.3883321033 0.6071689684
N 6.7014893581 0.3476094146 0.6297568787
C 6.3859836423 -0.9992575041 0.8326948605
O -3.7249569113 -4.5972293340 1.6128863003
O -4.8463201273 -0.1933666067 1.9790597102
O 6.1208337154 2.5362818857 0.3762622272
O 7.2455565123 -1.8716991890 0.7797777845
O 1.1407886896 2.6533726533 0.6063219715
C -5.6934127600 -2.8037590094 2.0407348857
C 8.0842859531 0.7160329905 0.3046306994
H -2.8985430976 1.3858998072 2.1683710260
H -0.5500821971 2.0372165163 1.9643845144
H 1.1158055270 -4.5119115177 1.4924034872
H -1.2766387362 -5.0806182408 1.5886693194
H 5.3593790729 -3.4104353647 1.2588367928
H 3.0074797636 -4.0275323577 1.5641451957
H 3.6698327225 3.0146094166 0.5148954053
H -6.1025426586 -2.9937710952 1.0451360813
H -6.2453037450 -1.9873059561 2.5052066127
H -5.7599649295 -3.7156343043 2.6361564253
H 8.1351799777 1.0444669621 -0.7382368735
H 8.3927688709 1.5446164711 0.9463066487
H 8.7138676664 -0.1584621682 0.4600662223
- Yes. You can see the scf process below. I think it does not matter.
1st
---------------------------------------
Cycle Energy DIIS error
---------------------------------------
1 -3308.7166863003 1.88e-02
2 -3312.7230965250 1.76e-02
3 -2589.1853031081 5.33e-02
4 -3325.5103987583 1.43e-02
5 -3343.7304260311 8.74e-03
6 -3222.3781036952 2.05e-02
7 -3346.7396485859 7.96e-03
8 -3271.9230556666 1.74e-02
9 -3352.3644604592 6.31e-03
10 -3360.0929774433 2.27e-03
11 -3358.7806090865 3.84e-03
12 -3360.9573964581 1.43e-03
13 -3361.1410695320 1.16e-03
14 -3361.1842692983 9.25e-04
15 -3361.3810656927 2.85e-04
16 -3361.3815588343 2.76e-04
17 -3361.3975523715 1.49e-04
18 -3361.4025507333 1.07e-04
19 -3361.4060355646 6.34e-05
20 -3361.4077836634 2.46e-05
21 -3361.4082129056 1.52e-05
22 -3361.4084648587 1.14e-05
23 -3361.4086473609 7.52e-06
24 -3361.4087768913 7.55e-06
25 -3361.4088293902 5.08e-06
26 -3361.4088539014 5.15e-06
27 -3361.4088679106 4.81e-06
28 -3361.4088861752 3.17e-06
29 -3361.4088967677 2.66e-06
30 -3361.4089080595 1.85e-06
31 -3361.4089135604 1.24e-06
32 -3361.4089134513 8.81e-07
33 -3361.4089153899 6.36e-07
34 -3361.4089157044 5.19e-07
35 -3361.4089162262 2.59e-07
36 -3361.4089163192 2.19e-07
37 -3361.4089163640 9.62e-08
38 -3361.4089163731 7.34e-08
39 -3361.4089163772 5.60e-08
40 -3361.4089163790 3.19e-08
41 -3361.4089163796 2.72e-08
42 -3361.4089163806 1.85e-08
43 -3361.4089163808 1.80e-08
44 -3361.4089163808 1.10e-08
45 -3361.4089163809 6.02e-09 Convergence criterion met
---------------------------------------
SCF time: CPU 35711.23s wall 4477.00s
SCF energy in the final basis set = -3361.4089163809
Total energy in the final basis set = -3361.4089163809
2nd
---------------------------------------
Cycle Energy DIIS error
---------------------------------------
1 -3342.5423211413 1.76e-02
2 -3325.0386060658 2.23e-02
3 -2936.8494466407 4.20e-02
4 -3396.1336797858 7.79e-03
5 -2837.6267018953 4.52e-02
6 -3284.7408735776 2.04e-02
7 -3396.1516994589 8.33e-03
8 -3303.4912048399 1.86e-02
9 -3409.4416390184 2.82e-03
10 -3409.1730501340 3.60e-03
11 -3411.1245485867 1.82e-03
12 -3411.5787758643 8.95e-04
13 -3411.6677545955 5.73e-04
14 -3411.7183628260 1.98e-04
15 -3411.7248476745 8.80e-05
16 -3411.7265220677 5.66e-05
17 -3411.7276448966 3.87e-05
18 -3411.7283545238 2.66e-05
19 -3411.7289177723 1.87e-05
20 -3411.7293905415 1.20e-05
21 -3411.7296761362 8.52e-06
22 -3411.7298410293 7.06e-06
23 -3411.7299131911 5.06e-06
24 -3411.7299962495 3.86e-06
25 -3411.7300343005 3.00e-06
26 -3411.7300479124 1.70e-06
27 -3411.7300537839 1.13e-06
28 -3411.7300572530 9.27e-07
29 -3411.7300578636 6.22e-07
30 -3411.7300584838 5.34e-07
31 -3411.7300586434 2.48e-07
32 -3411.7300587583 2.36e-07
33 -3411.7300588403 1.80e-07
34 -3411.7300588875 1.51e-07
35 -3411.7300589281 1.07e-07
36 -3411.7300589603 6.99e-08
37 -3411.7300589719 4.12e-08
38 -3411.7300589745 3.22e-08
39 -3411.7300589756 1.77e-08
40 -3411.7300589761 1.04e-08
41 -3411.7300589762 6.69e-09 Convergence criterion met
---------------------------------------
SCF time: CPU 29435.15s wall 3691.00s
SCF energy in the final basis set = -3411.7300589762
Total energy in the final basis set = -3411.7300589762
- However, I’m testing the other functionals like CAM-B3LYP and WB97XD and the jobs are proceeding well. Actually, I’m trying to select the proper functionals among the functionals sets which are designed to consider the non-bonded interactions as you know. And I think dispersion should be contained as the system must remain the separation of the chromophores although M06-2X looks bad. What do you think of?
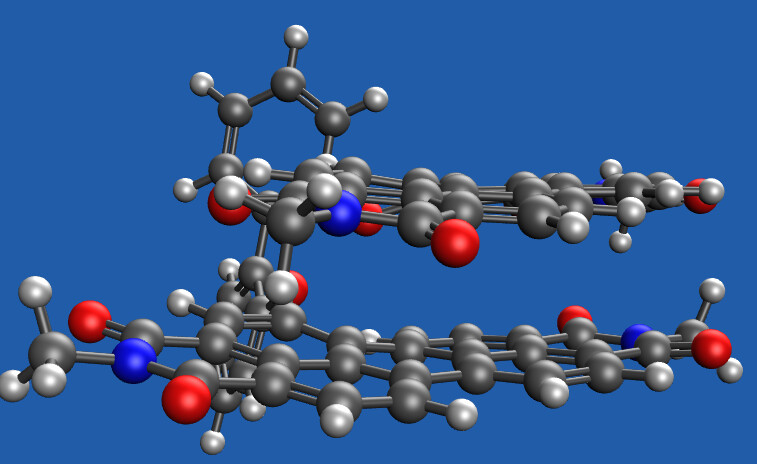
- The starting geometry indeed looks reasonable.
- Okay, SCF is converged without issues.
- For troubleshooting purposes I recommend to run it without the D correction. This will test the hypothesis of whether it is the cause of the problem. This is completely independent from whatever interaction benchmarks.
I would also suggest that based on slow SCF convergence that you probably need to tighten the thresholds for a molecule of this size (Q-Chem defaults are not appropriate). Suggest THRESH=12.
Furthermore, M06-2X-D3 is a bit of an outlier that significantly overbinds large van der Waals complexes, which would be consistent with the behavior that you observe. See here:
so this may be an artifact of just that particular functional.
As you recommanded, I run it without the dispersion option. I track all geometries for each SCF step and it does not show any problems.
However, it is strange that final SCF energy is higher than the initial energy.
Can you give me an advice for it?
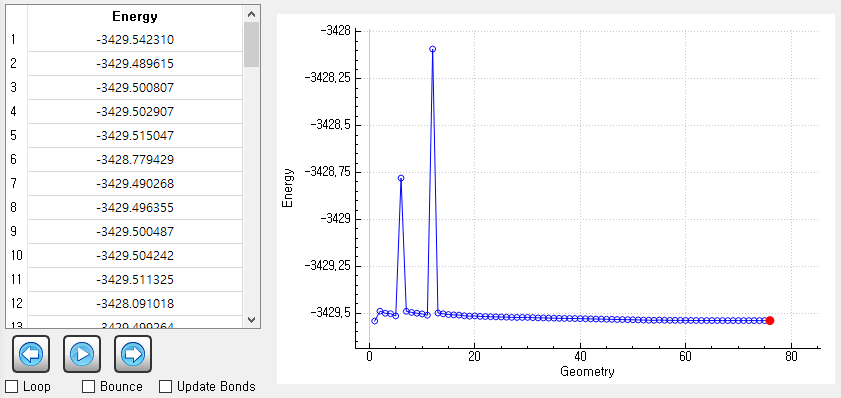
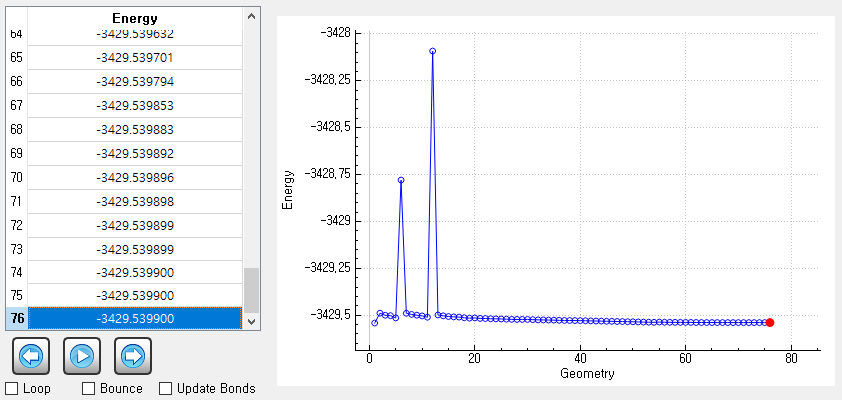
Your system has many degrees of freedom. It is possible that the geometry optimization procedure found another rotamer, for example. One should inspect closely how the geometry changes and whether the initial geometry is qualitatively different from the optimized one.
But so far it looks like indeed the D correction was the issue with this particular functional.
Again, I think the issue is not with all +D corrections in general but rather with this specific functional, which seems to overbind and in this case that might manifest as pushing the two pi systems together.
If you are running a recent version of Q-Chem (6.0 or higher, I think) then another consideration is that you are using a new geometry optimizer that does not employ a very good initial guess Hessian. That’s an oversight that’s getting fixed in Q-Chem 6.2 but for 6.0 and 6.0 you can specify
$geom_opt
initial_hessian model
$end
to get a better initial guess for the BFGS Hessian.