The structure and chemical reactions of molecules under high pressures has become a fascinating research topic [1-3], thanks to the development of high pressure technology and instruments. The experimental progress in high-pressure chemistry has posed a question to theoretical chemists: how can we model molecular systems under pressure?
In the new release of Q-Chem v5.4, a module called DISTORT (developed by Prof. Tim Stauch and coworkers) is provided to study the geometry and electronic structures, as well as the dynamics of molecules under user-defined pressures. Three theoretical models of external pressure application are offered:
- the Hydrostatic Compression Force Field (HCFF) model [4];
- the eXtended Hydrostatic Compression Force Field (XHCFF) model [5]; and
- the Gaussians On Surface Tesserae Simulate HYdrostatic Pressure (GOSTSHYP) model [6].
A $rem variable “distort” is used to turn on the calculation under pressure. An input section “$distort” is used to the parameters in such a calculation (see the user’s manual of Q-Chem 5.4 for details). For all methods in DISTORT the energy gradients are available (e. g., in a force calculation). Two common types of calculations are now supported: geometry optimization and ab initio molecular dynamics (AIMD), as illustrated in the following examples:
Example 1. Calculating the bond lengths and total energies of H2+ at different pressures using the HCFF model.
Q-Chem Input for the external pressure = 1000 MPa = 10000 bar (comments are put after the “!” sign and should be removed in case qchem does not run properly):
$molecule
1 2
H 0.0 0.0 0.0
H 0.0 0.0 1.058
$end$rem
jobtype opt ! this is a geometry optimization calculation
method hf
basis cc-pVTZ
distort true ! pressure is applied onto the molecule
$end$distort
model hcff ! using the HCFF model
pressure 1000.0 ! pressure = 1000 MPa = 10000 bar
scaling 1.0 ! the scaling factor of the atomic van der Waals radii (1.0 means no scaling)
npoints_heavy 590 ! the number of tessellation points per non-hydrogen atom
npoints_hydrogen 590 ! the number of tessellation points per hydrogen atom
$end
Usually one can just use the recommended settings for “scaling”, “npoints_heavy” and “npoints_hydrogen” in the qchem manual. However, if the user decides to use a lower number of tessellation points per atom than the default (110), the convergence of the calculated results should be checked. The value of “scaling” should be increased from the default (1.0 for X-HCFF and 1.2 for the other models) when modeling a chemical complex to make sure that the complex is placed inside a single cavity.
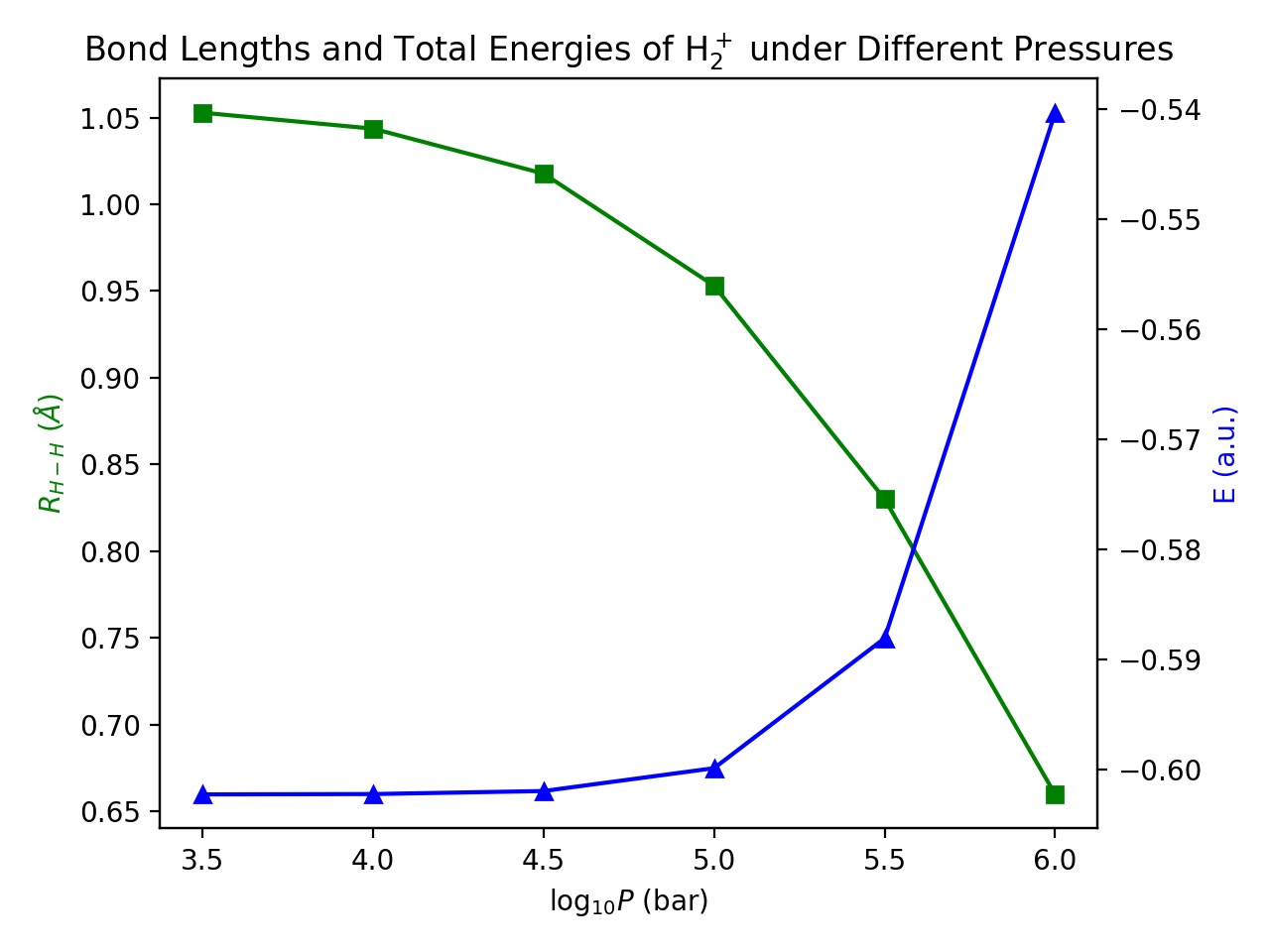
Vary the pressure from 316.2 MPa (=10 3.5 bar) to 100000 MPa (=106.0 bar), The calculated bond lengths and total energies can be plotted as
Example 2. Geometry optimization of a Diels-Alder reaction system (cyclopentadiene and ethylene) under pressure using the GOSTSHYP model to find the transition pressure.
Q-Chem Input for the external pressure = 35000 MPa = 35 GPa (comments are put after the “!” sign and should be removed in case qchem does not run properly):
$molecule
0 1
C -0.78854 -0.68781 -1.31508
C -0.78854 0.68781 -1.31508
C 0.39404 -1.14676 -0.77519
C -0.26584 -0.66530 2.12986
C -0.26584 0.66530 2.12986
C 0.39404 1.14676 -0.77519
C 1.25986 0.00000 -0.38117
H 2.21173 0.00000 -0.95202
H 1.45683 0.00000 0.71144
H -1.58727 1.31951 -1.68000
H -1.58727 -1.31951 -1.68000
H 0.65908 -1.21172 2.28266
H -1.19088 -1.21171 1.97767
H -1.19088 1.21171 1.97767
H 0.65908 1.21172 2.28266
H 0.64783 -2.19216 -0.65967
H 0.64783 2.19216 -0.65967
$end$rem
symmetry off
sym_ignore true
jobtype opt
method pbe
basis cc-pvdz
max_scf_cycles 500
gen_scfman true
use_libqints true
distort true ! pressure is applied onto the molecule
scf_final_print 1 ! the energy contribution of the pressure is printed after SCF convergence
$end
$distort
model gostshyp ! the GOSTSHYP model is used
pressure 35000 !pressure = 35000 MPa = 35 GPa
npoints_heavy 302
npoints_hydrogen 302
scaling 1.8 ! non-default scaling factor is used
$end
Here, a non-default scaling factor = 1.8 is used to ensure that the entire molecular system is in a single cavity [6].
Vary the pressure from 35 GPa to 45 Gpa, the calculated C—C distances (d) and energy contributions (Ep) from the pressure can be plotted as
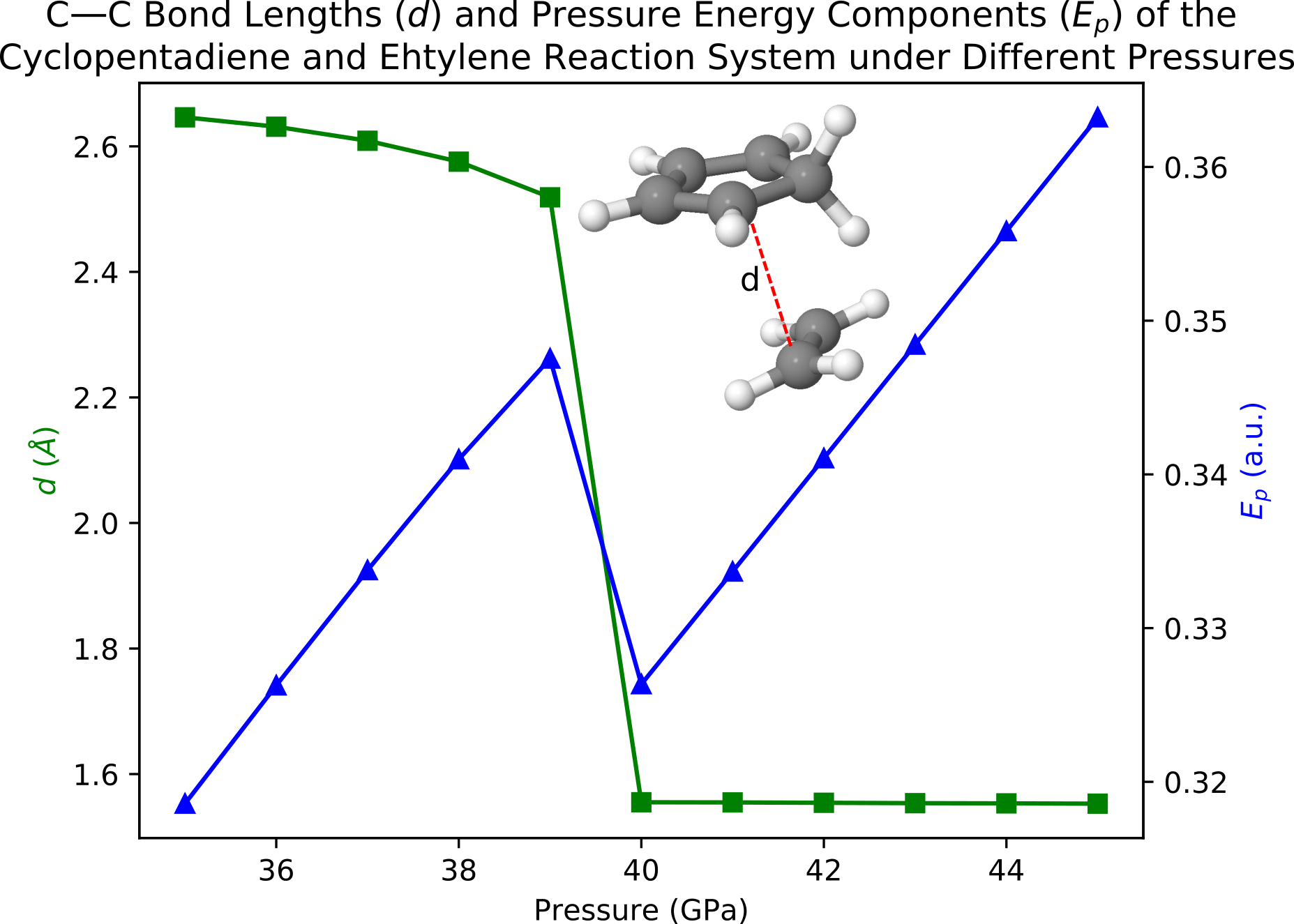
One can easily see that both d and Ep show a sudden decrease between pressure = 39 and 40 GPa, which indicates that a Diels-Alder reaction is happening if we apply a pressure of 40 GPa on this system. In the next example, we confirm the happening of the Diels-Alder reaction through AIMD simulations.
Example 3. AIMD simulation of a Diels-Alder reaction system (cyclopentadiene and ethylene) under pressure using the GOSTSHYP model.
Q-Chem Input for the external pressure = 40000 MPa = 40 GPa (comments are put after the “!” sign and should be removed in case qchem does not run properly):
$molecule
0 1
C -0.78854 -0.68781 -1.31508
C -0.78854 0.68781 -1.31508
C 0.39404 -1.14676 -0.77519
C -0.26584 -0.66530 2.12986
C -0.26584 0.66530 2.12986
C 0.39404 1.14676 -0.77519
C 1.25986 0.00000 -0.38117
H 2.21173 0.00000 -0.95202
H 1.45683 0.00000 0.71144
H -1.58727 1.31951 -1.68000
H -1.58727 -1.31951 -1.68000
H 0.65908 -1.21172 2.28266
H -1.19088 -1.21171 1.97767
H -1.19088 1.21171 1.97767
H 0.65908 1.21172 2.28266
H 0.64783 -2.19216 -0.65967
H 0.64783 2.19216 -0.65967
$end$rem
symmetry off
sym_ignore true
jobtype aimd ! this is an AIMD calculation
aimd_method bomd
method pbe
basis cc-pvdz
max_scf_cycles 500
gen_scfman true
use_libqints true
distort true ! pressure is applied onto the molecules
mem_total 24000
mem_static 2000
TIME_STEP 20
AIMD_STEPS 2000
AIMD_INIT_VELOC thermal
AIMD_TEMP 298
$end$distort
model gostshyp ! the GOSTSHYP model is used
pressure 40000 ! pressure = 40000 MPa = 40 GPa
npoints_heavy 302
npoints_hydrogen 302
scaling 1.8
$end
The resulted time-dependent distance between two C atoms in the reactants of a representative AIMD simulation can be shown in the following plot as:
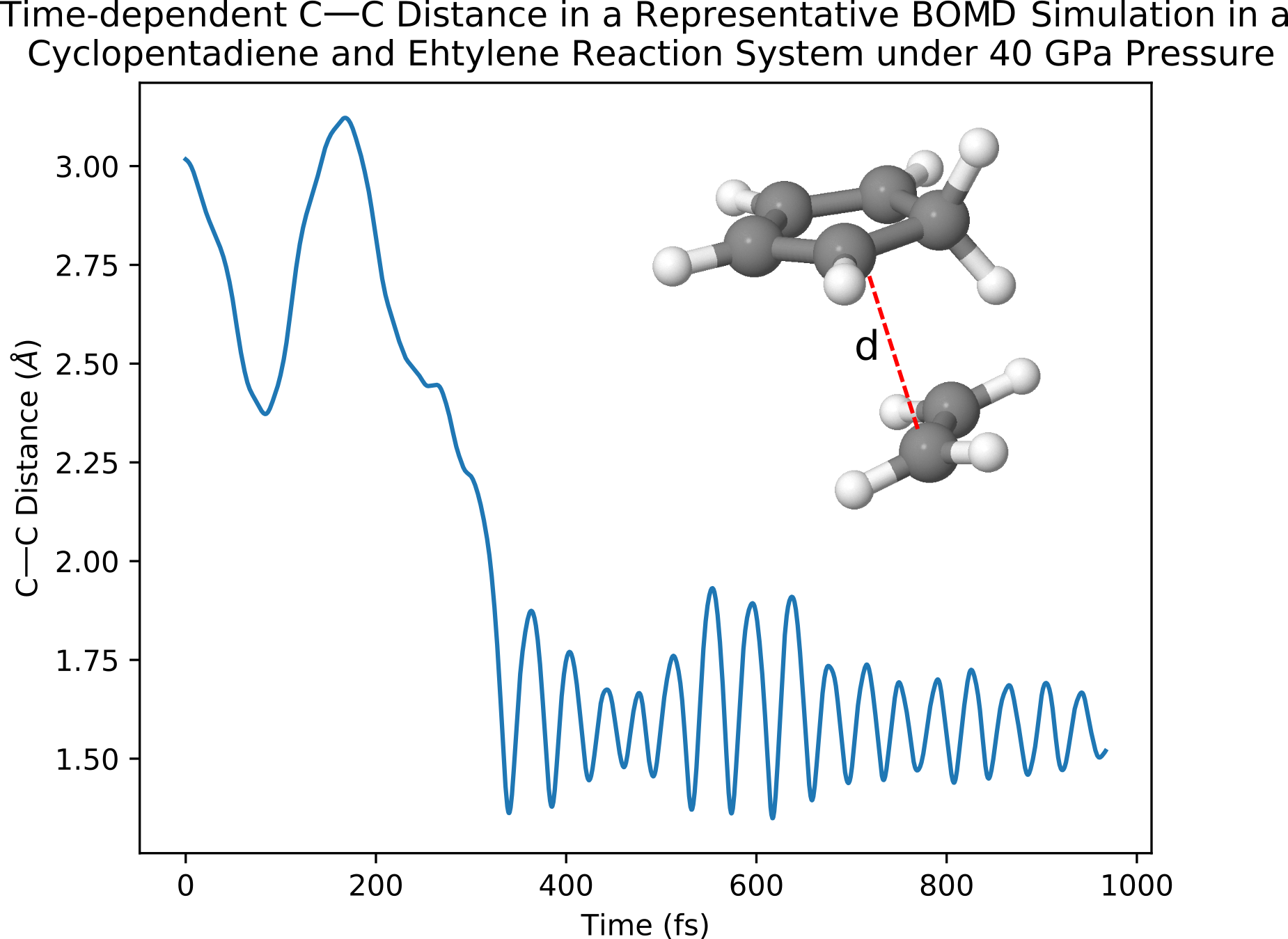
Here one can see that after 200 fs the distance between the two C atoms (as indicated in the figure) drops from > 2.25 angstrom to ~ 1.6 angstrom, which indicates the happening of the Diels-Alder reaction. Note: this calculation may take some time to finish. Furthermore, because of the random nature of AIMD calculations, one may not obtain the same result in the figure above and need to run multiple simulations in order to find a representative trajectory indicating the happening of the Diels-Alder reaction.
Reference:
- P. F. McMillan, Chem. Soc. Rev. 35, 855 (2006).
- W. Grochala et al., Angew. Chem. Int. Ed. 46, 3620 (2007).
- V. Schettino and R. Bini, Chem. Soc. Rev. 36, 869 (2007).
- T. Stauch, R. Chakraborty and M. Head-Gordon, ChemPhysChem 20, 2742 (2019).
- T. Stauch, J. Chem. Phys. 153, 134503 (2020).
- M. Scheurer et al., J. Chem. Theory Comput. 17, 583 (2021).