Hello everyone!
I’ve been trying to perform geometry optimization of T1 state of a molecule in solution (ACN). Depending on the options used I’ve been getting two distinctly different geometries and some perplexing results, which I’m hoping you’ll help me understand.
I use two-job inputs, as in the first job I generate orbitals in desired, but user-defined basis set (DZP, since it’s not available). The second part of the input is as follows:
$molecule
read
$end
$rem
jobtype opt
method camb3lyp
dft_d d3_bj
basis gen
purecart 11
PRINT_GENERAL_BASIS false
gen_scfman false
MAX_SCF_CYCLES 500
SCF_GUESS read
SCF_CONVERGENCE 9
scf_final_print true
MEM_STATIC 2000
MEM_TOTAL 80000
GEOM_OPT_MAX_CYCLES 200
CIS_N_ROOTS 3
CIS_TRIPLETS true
CIS_SINGLETS false
CIS_RELAXED_DENSITY true
CIS_STATE_DERIV 1
RPA false
MOLDEN_FORMAT true
IQMOL_FCHK true
solvent_method pcm
$end
$pcm
NonEquilibrium true
Theory iefpcm
StateSpecific perturb
$end
$solvent
solventname acetonitrile
$end
As I’ve mentioned, I have been trying different variations of the $pcm section, which are summed up in the table:
| NonEquilibrium | Theory | StateSpecific | |
|---|---|---|---|
| orig | TRUE | iefpcm | perturb |
| v1 | - | - | - |
| v2 | FALSE | cpcm | - |
| v3 | FALSE | iefpcm | perturb |
| v4 | TRUE | cpcm | - |
| v5 | FALSE | iefpcm | perturb |
| v6 | FALSE | cpcm | perturb |
| v7 | - | iefpcm | perturb |
| v8 | TRUE | iefpcm | - |
| v9 | - | iefpcm | - |
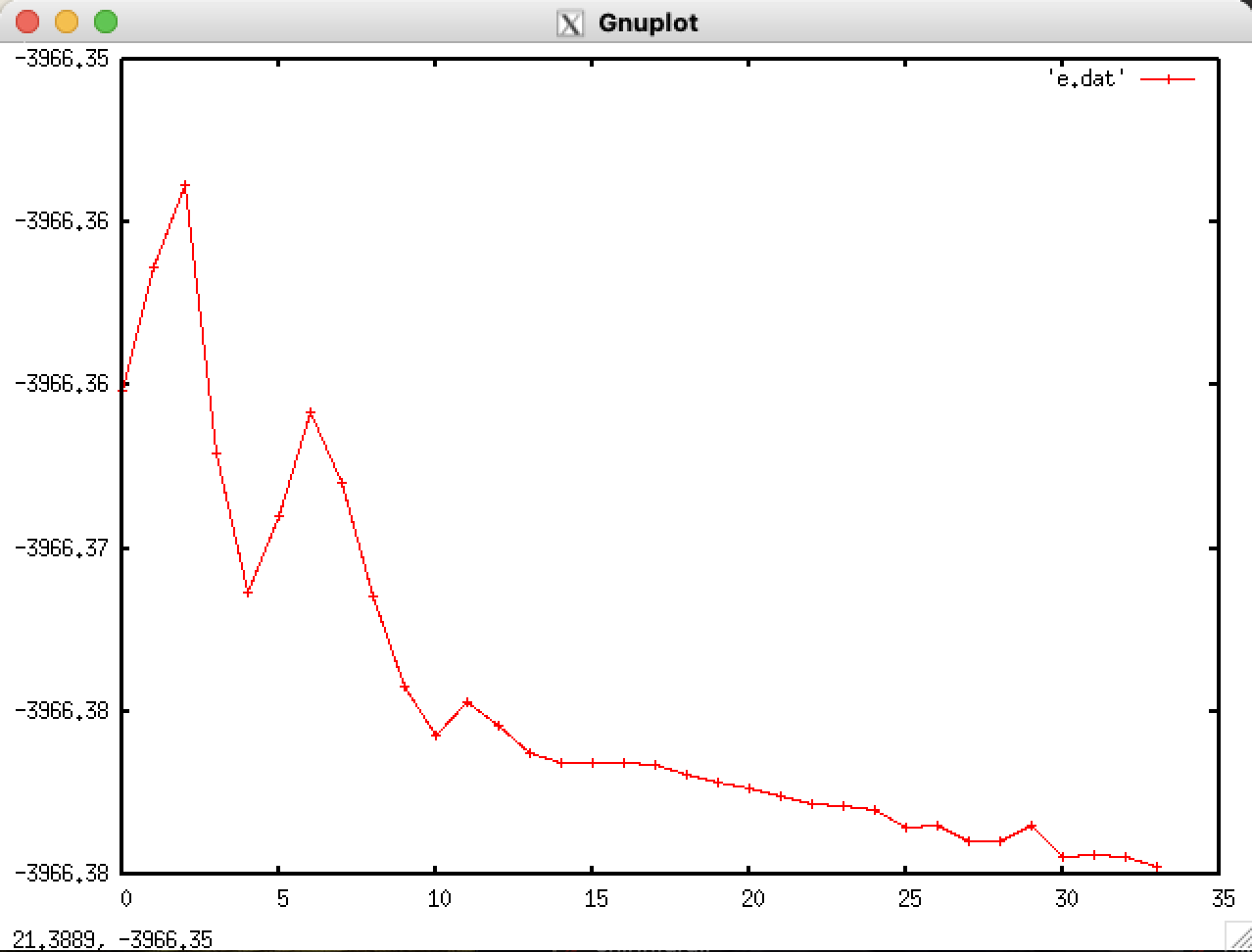
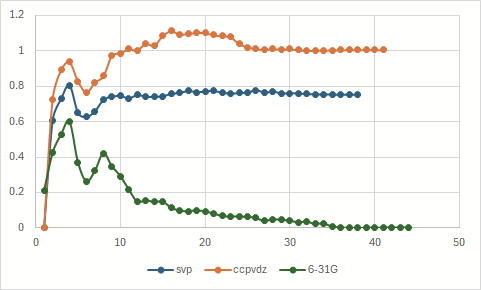
Energy convergence during the optimizations:
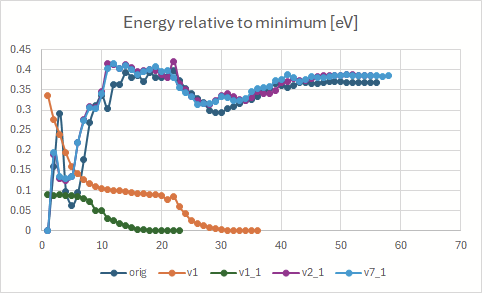
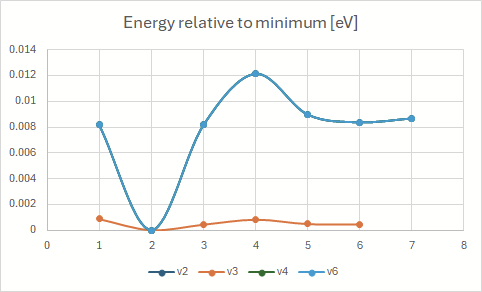
The energies here are the ones given after each step, for example:
Step 5 :
Energy is -3967.0220022392
Starting point was the geometry obtained using the “orig” input file, or, if “_1” is present, a different one (usually the geometry obtained in v1 job).
In the second picture jobs v2, v4 and v6 resulted in exactly same energies.
Some jobs aren’t depicted above. v5, v7 and v8 indicated an already converged starting geometry (convergence in one step). v9 resulted in en error - SCF did not converge.
And here are my questions:
- Why does energy go up during the optimization?
Please refer to the pictures. As far as I know, the geometry should go down during optimization - but in some cases here it doesn’t, yet the job finished as successful. Why is that?
- How should I actually approach the geometry optimisation?
I’m not sure which is the correct approach here, since the geometry obtained in all the jobs except v1 results in the same structure, however the v1 job gives a structure that is virtually the same as one obtained using a different programme (also in pcm), but it’s also the same as the structure I get in vacuum.
- What do the options “NonEquilibrium” and “StateSpecific” actually do?
Comparing jobs v2 and v4 as well as orig and v3 shows that neither the final structures and energies depend on the “NonEquilibrium” value. Similar situation seems to appear for job orig and v5 - same structure, same energy. However, if I remove the “NonEquilibrium” option, the scf doesn’t converge - compare v8 and v9.
I’ll be grateful for any input on that matter.
Best regards,
Joanna