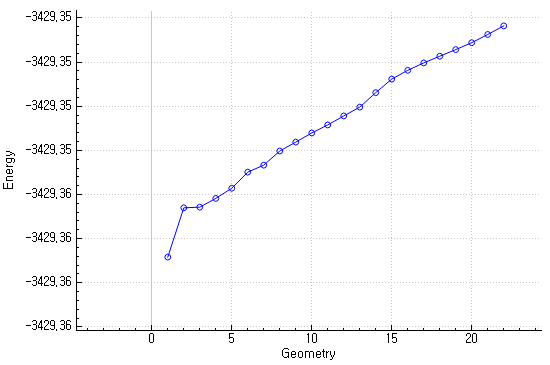
However, the energy change looks strange.
I think the energy should be decreased to be converged to the specific minima but the energy is increasing now still running the job.
Is it right? Or if it’s wrong, what can I do to overcome this situation?
If this graph was plotted with IQmol then this is likely the ground state energy and it is not supposed to decrease with an excited state optimization.
You can open the Q-Chem output with any text editor and check if, with every step, the energy of the first excited state decreases, as you requested in your input.
Yes. That graph is plotted with IQmol so I check the S1 state energy from Q-Chem output file.
Here are the results.
Total energy for state 1: -3429.25721256 au
Total energy for state 1: -3429.25174348 au
Total energy for state 1: -3429.25207843 au
Total energy for state 1: -3429.25199076 au
Total energy for state 1: -3429.25314142 au
Total energy for state 1: -3429.25280396 au
Total energy for state 1: -3429.25262238 au
Total energy for state 1: -3429.25209577 au
Total energy for state 1: -3429.25191504 au
Total energy for state 1: -3429.25161187 au
Total energy for state 1: -3429.25142120 au
Total energy for state 1: -3429.25121847 au
Total energy for state 1: -3429.24909165 au
Total energy for state 1: -3429.25047243 au
Total energy for state 1: -3429.25000709 au
Total energy for state 1: -3429.24971131 au
Total energy for state 1: -3429.24949459 au
Total energy for state 1: -3429.24930631 au
Total energy for state 1: -3429.24914879 au
Total energy for state 1: -3429.24898417 au
Total energy for state 1: -3429.24879500 au
Total energy for state 1: -3429.24860636 au
Total energy for state 1: -3429.24844171 au
Total energy for state 1: -3429.24828599 au
Total energy for state 1: -3429.24809977 au
Total energy for state 1: -3429.24798074 au
Total energy for state 1: -3429.24783573 au
Total energy for state 1: -3429.24772025 au
Total energy for state 1: -3429.24758991 au
Total energy for state 1: -3429.24751172 au
Total energy for state 1: -3429.24742194 au
Total energy for state 1: -3429.24736116 au
Probably means your optimization is oscillating. Check for nearby electronic states (i.e., how much higher in energy is excited state 2?). May be that you are unable to find an adiabatic state (as specified using CIS_STATE_DERIV) of consistent electronic character. If so, that’s a bug in the Born-Oppenheimer approximation, not a bug in the code
Its possible that there is no proper excited state minima but an MECP with S0 instead that the optimizer may be trying to lead you to - how close is S0 in energy? Try running a frequency at the lowest energy point you can find and look for modes with massive frequencies ([2308.06475] Fantastical Excited State Optimized Structures and Where to Find Them)