Dear all,
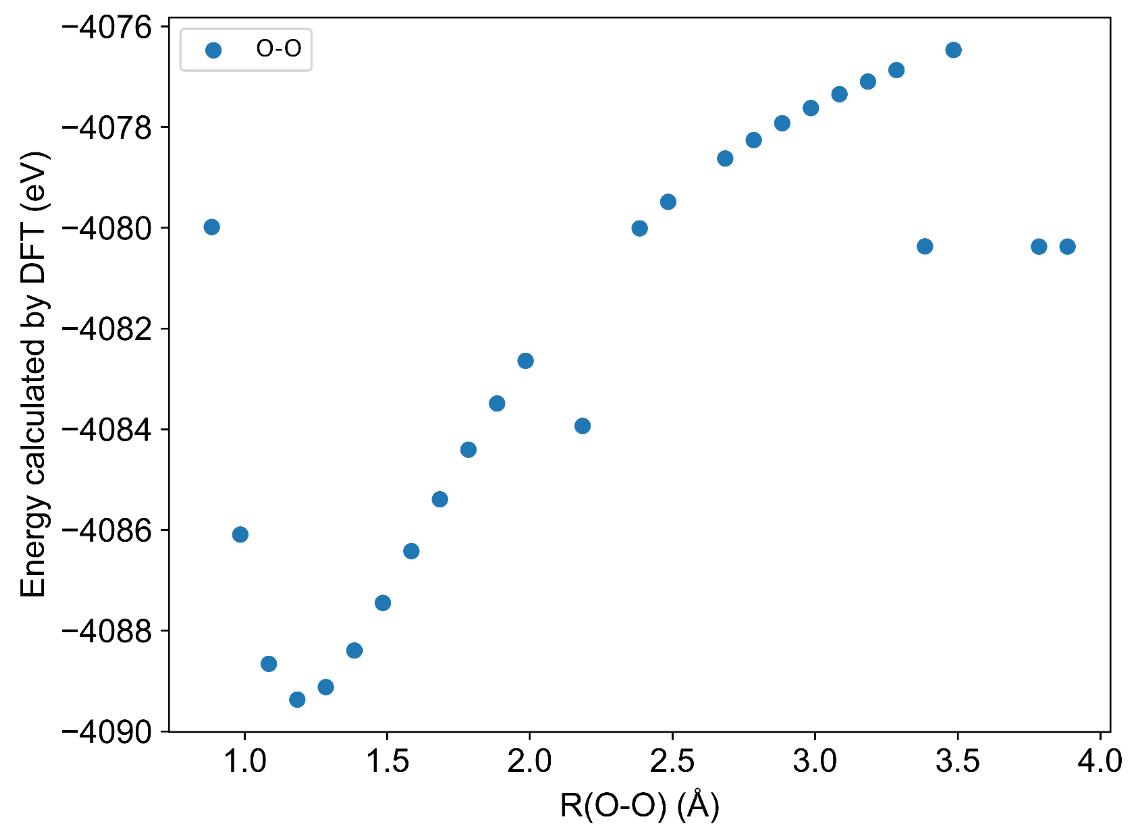
I am trying to verify the spin selection for the hydrogen combustion system with the wB97M-V method. For example, I choose three ways to calculate the energy with the wB97M-V method and cc-pVTZ basis set: spin=1, spin=3, and guess=mix, and the lowest energy was chosen to draw the dissociation curve. The dissociation curve is not smooth. Here is one input of my input files:
spin =1 or 3:
$molecules
0 1(3)
O 11.9573 7.97905 12.3234
O 12.4226 6.78293 12.3816
$end
$rem
JOBTYPE FORCE
MEM_TOTAL 24000
METHOD wB97M-V
BASIS cc-pVTZ
UNRESTRICTED true
SYMMETRY false
SYM_IGNORE true
$end
After that, I adopted the INTERNAL_STABILITY = TRUE in my input to perform internal stability analysis and generate the corrected MOs. But the SCF failed to converge.
Can someone explain why is this happening? Is this keyword( INTERNAL_STABILITY) equivalent to stable=opt in Gaussian?
Looks like there’s a lower-energy solution to which your calculation converges intermittently, which is not unusual for highly stretched bonds. The way that I would generate such a curve is to start from the equilibrium distant (or from R=2 A in your case, since the curve is well-behaved up to that point) and then compute the longer bond lengths by reading in the MOs from the previous (shorter R) calculation and using the maximum overlap method to attempt to find a solution that looks like the one at the shorter R. I’ll paste an example below, but a couple of other things about your input file that are strange are:
(1) why set JOBTYPE=FORCE just to compute a potential energy scan? Do you need the gradient?
(2) SCF_GUESS = GWH is an odd choice, was there a reason?
(3) Your memory settings much higher than needed for such a small molecule
$rem
method wB97M-V
basis cc-pVTZ
unrestricted true
scf_guess read
mom_method imom ! usually works better than original MOM
mom_start 1 ! begin MOM immediately on 1st SCF cycle
$end
@@@
$comment
Finally, R = 2.50 A
$end
$molecule
0 1
O
O 1 2.50
$end
$rem
method wB97M-V
basis cc-pVTZ
unrestricted true
scf_guess read
mom_method imom ! usually works better than original MOM
mom_start 1 ! begin MOM immediately on 1st SCF cycle
$end
Using the approach above, starting from R=2.0 A in 0.25 A increments, I get the following smooth potential energy curve for the singlet state (which is not the ground state for O2…)
Thanks for your reply and I’m very sorry for not writing clearly that caused your question. Here are my answers:
(1) Actually I need to get energy and atomic forces to make my dataset, and getting the oxygen dissociation curve or hydrogen dissociation curve is one part of the tasks to verify the correctness of the method. Beacuse the dataset is bulit with one-bodys and two-bodys, the smooth of the energy surface need to be verified.
(2) SCF _GUESS = GWH is refer to Example 4.60, and the input in this example is for an unrestricted HF calculation on H2 in the dissociation limit.
(3) I will lower the value of memory settings, and thanks for your warning.
Thanks for providing the example! I want to know that how to set the SCF_GUESS_MIX, which is corresponding to what proportion of the alpha LUMO mixed with the alpha HOMO. In your example, the set scf_guess_mix was set to 5.
The examples in the manual are just that: examples. They should not necessarily be taken as the definitive way to do a certain calculation, just a way.
SCF_GUESS_MIX = 5 mixes 50% HOMO and 50% LUMO, I think, but I chose that sort of instinctively and your value of 2 might have worked also. It’s likely not that sensitive so long as the fraction isn’t tiny. The purpose is just to break spin symmetry in the guess, which let’s the system unrestrict if that lowers the energy. (A spin-symmetric guess can be a saddle point that won’t unrestrict unless you nudge it off of that point, which you do by mixing alpha and beta.)