Hello,
I am trying to do a CIS calculation on a dimeric system containing total of 164 atoms including 8 bromine atoms. My aim is to study triplet energy transfer. The $rem variables used are
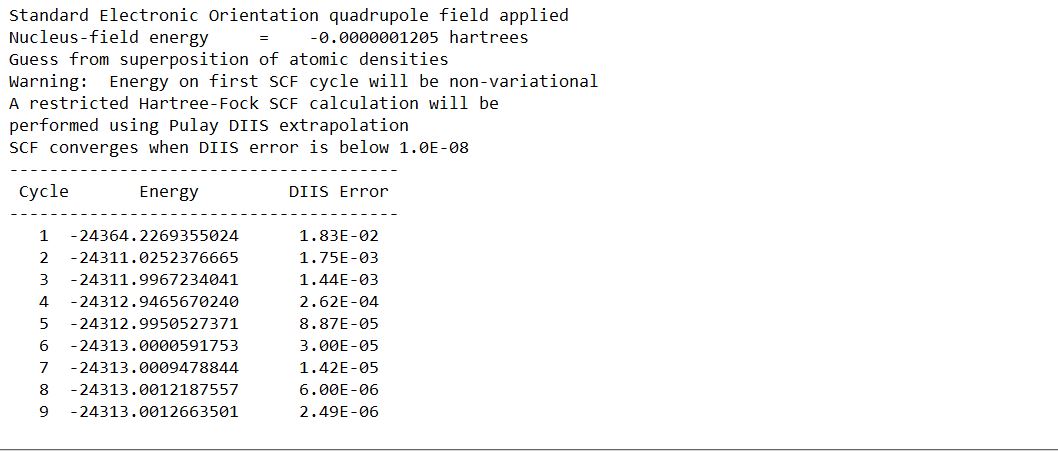
But the calculation is getting terminated without showing any error. This means it seems like the calculation has not finished but no more lines are getting added to the output file. Can someone explain why is this happening?
The last part of the output file looks like
I doubt this is causing your particular problem, but I was unaware of “METHOD = CIS” as an option, and in fact when I try it on a small test molecule I get a ground-state Hartree-Fock calculation but no excited states at all. The proper nomenclature for CIS in Q-Chem is METHOD = HF followed by CIS_N_ROOTS = … and then (optionally) the CIS_SINGLETS and CIS_TRIPLETS variables.
Is there a reason you’ve set MEM_STATIC=1500? I would check that as well.
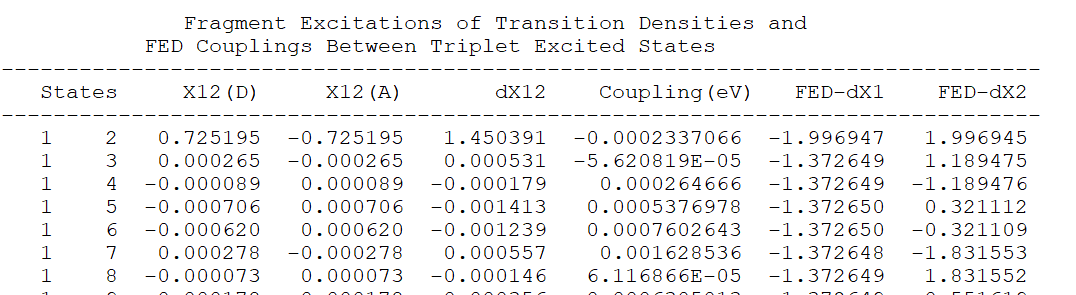
Hello, I also used this input file to calculate the triplet energy transfer, but the output file does not seem to have the energy transfer between T1-T1. Does your calculated result file have energy transfer between T1-T1?Thank you.