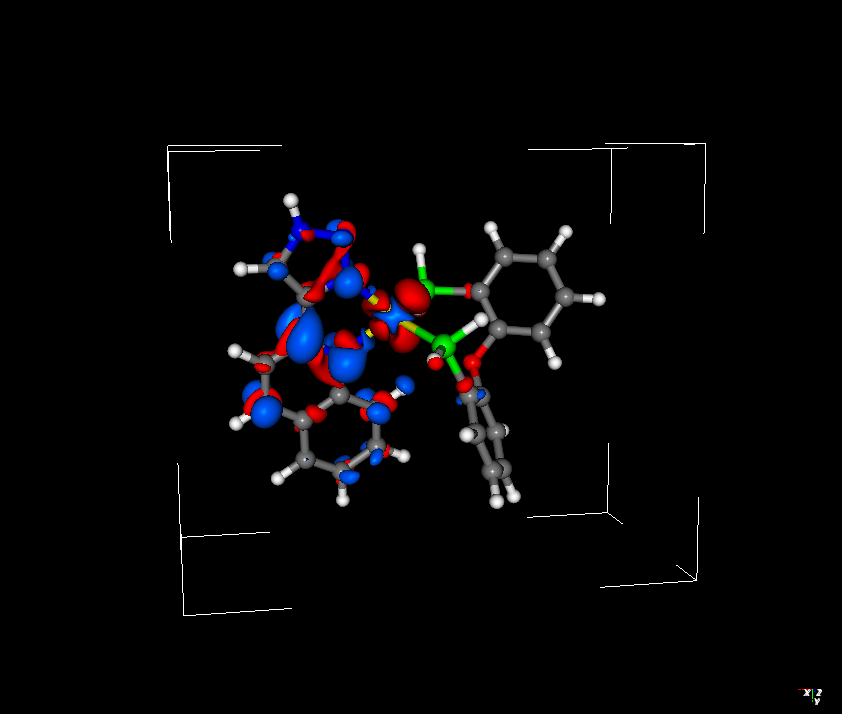
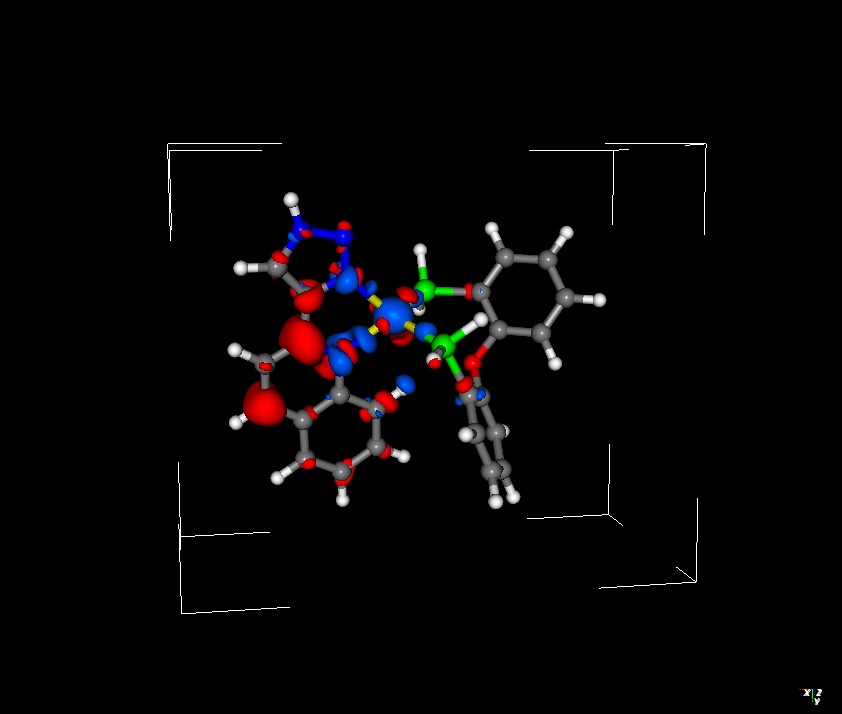
I have been trying to compare how ROKS and TDDFT describe the lowest singlet excited state of a Cu(I) complex. It is well known that the S1 state of such a complex is of MLCT nature. I wanted to compare the difference densities obtained by TDDFT and ROKS methods, and while the TDDFT difference density looks reasonable, the ROKS difference density is somewhat bizarre - both in terms of the shape (the density around copper doesn’t even resemble d orbitals), and the sign (blue colour corresponds to increase in electron density, and red - decrease. Since S1 is MLCT state, the electron density should shift from Cu to the diimine ligand - ROKS difference density suggests the opposite direction).
It would be better to show your Q-Chem input files of two jobs. Especially for S1 ROKS, this is tricky and thus it is important to know whether your calculation is reasonable.
Oh, right, sorry. Here is the input for ROKS job (I’m omitting the $basis section at the end):
$molecule
1 1
C -0.0861094 0.2614760 4.4284010
C 0.5096588 0.3757065 3.1816150
C 1.0496294 1.5837268 2.7359403
C 1.0015825 2.6871167 3.5861737
C 0.4124758 2.5873670 4.8404721
C -0.1333138 1.3783079 5.2546878
O 0.6338685 -0.6873111 2.3114184
C -0.3459637 -1.6608830 2.2800812
C -1.5413836 -1.4062650 1.6026722
C -2.4754908 -2.4388502 1.5126745
C -2.2186179 -3.6821212 2.0770633
C -1.0196214 -3.9120354 2.7408295
C -0.0762404 -2.8967669 2.8442780
P -1.7757750 0.2056633 0.7680290
Cu -0.0099175 1.0374774 -0.4300330
N -0.4956207 2.3714615 -1.9491262
N -0.8717911 3.5984554 -2.0622932
N -0.9896102 3.8322542 -3.3630775
C -0.6887778 2.7479908 -4.1100858
C -0.3648591 1.7919690 -3.1788684
H -1.2783050 4.7547966 -3.6728069
C 0.0612041 0.3934239 -3.2684041
N 0.3547329 -0.1762412 -2.1124075
C 0.7603755 -1.4765272 -2.0871350
C 0.8556498 -2.2408204 -3.2793213
C 0.5337349 -1.6016071 -4.4985454
C 0.1432029 -0.2921021 -4.4993900
C 1.2637142 -3.5940535 -3.1972013
C 1.5636959 -4.1527215 -1.9842185
C 1.4784912 -3.3819513 -0.8021984
C 1.0905607 -2.0705839 -0.8484187
P 1.6202624 1.6680347 1.0064964
H 1.0327142 -1.4617953 0.0513896
H 1.7249879 -3.8376432 0.1562885
H 1.8749591 -5.1949944 -1.9250534
H 1.3340135 -4.1802782 -4.1136924
H 0.6002212 -2.1621707 -5.4315054
H -0.1095659 0.2179378 -5.4271557
H -0.7256958 2.7520097 -5.1932227
H -3.0269040 -0.0460659 0.1565302
H -2.2500413 1.0089958 1.8356931
H 0.8744906 -3.0470020 3.3546538
H -0.8161283 -4.8867822 3.1831365
H -2.9599008 -4.4759748 1.9944196
H -3.4158473 -2.2722399 0.9870773
H 1.4191255 3.6397364 3.2594399
H 0.3785236 3.4572959 5.4948016
H -0.5972867 1.2965012 6.2371540
H -0.5036939 -0.6913384 4.7499609
H 2.8470419 0.9599768 1.0487696
H 2.1497972 2.9800872 0.9947822
$end
This is second part if the S1 tddft calculation, in the first part I only generate the GS orbitals in my basis set. The basis set is the same in all my calculations.
Have you tried looking at the overlap with the ground state? You accomplish this by using the TRANS_MOM_SAVE = 1 $rem variable for a previous ground state calculation and then TRANS_MOM_READ = 2 for the ROKS calculation. Large overlaps are bad news. I believe the GS calculation needs to be unrestricted for TRANS_MOM_SAVE to work…
Sorry, I mixed up two geometries. I get zero for the perpendicular geometry of the system, and here I posted data for the 95 degree dihedral. I’m sorry for the confusion.
So in the proper case the overlap was 0.020555
Output of the ROKS calculation:
Standard Electronic Orientation quadrupole field applied
Nucleus-field energy = 0.0000000649 hartrees
Guess MOs from SCF MO coefficient file
Reading MOs from coefficient file
Reading MOs from coefficient file
Long-range K will be added via erf
Coulomb attenuation parameter = 0.33 bohr**(-1)
A restricted hybrid HF-DFT SCF calculation will be
performed using Pulay DIIS extrapolation
Exchange: 0.1900 Hartree-Fock + 0.3500 B88 + 0.4600 muB88 + LR-HF
Correlation: 0.1900 VWN5 + 0.8100 LYP
Using SG-1 standard quadrature grid
Dispersion: Grimme D3
SCF converges when DIIS error is below 1.0E-09
Using Q-Chem read-in guess as SCF_GUESS READ specified.
ROKS singly occupied orbitals: (125,126)
using 24 threads for integral computing
-------------------------------------------------------
OpenMP Integral computing Module
Release: version 1.0, May 2013, Q-Chem Inc. Pittsburgh
-------------------------------------------------------
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.95 sec.
Timing for roks_build(): 349.88 sec.
---------------------------------------
Cycle Energy DIIS Error
---------------------------------------
1 -3505.0703927334 1.25E-02
Det[Ca.S.C0] = -0.760754
Det[Cb.S.C0] = -0.005371
<S1|S0> = 0.005778
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 376.18 sec.
2 -3502.1006273839 9.13E-03
Det[Ca.S.C0] = -0.879849
Det[Cb.S.C0] = -0.005003
<S1|S0> = 0.006226
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.92 sec.
Timing for roks_build(): 364.24 sec.
3 -3504.0822708645 4.59E-03
Det[Ca.S.C0] = -0.936623
Det[Cb.S.C0] = -0.004648
<S1|S0> = 0.006157
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 358.29 sec.
4 -3504.7295149752 2.56E-03
Det[Ca.S.C0] = 0.967282
Det[Cb.S.C0] = 0.006658
<S1|S0> = 0.009108
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.92 sec.
Timing for roks_build(): 350.97 sec.
5 -3504.9883217290 1.32E-03
Det[Ca.S.C0] = 0.974097
Det[Cb.S.C0] = 0.005653
<S1|S0> = 0.007787
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 4.00 sec.
Timing for roks_build(): 350.68 sec.
6 -3505.0346733375 9.74E-04
Det[Ca.S.C0] = -0.978704
Det[Cb.S.C0] = -0.006548
<S1|S0> = 0.009064
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.97 sec.
Timing for roks_build(): 348.45 sec.
7 -3505.0633345492 6.93E-04
Det[Ca.S.C0] = 0.981559
Det[Cb.S.C0] = 0.007090
<S1|S0> = 0.009841
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 348.70 sec.
8 -3505.0793772928 4.80E-04
Det[Ca.S.C0] = 0.983893
Det[Cb.S.C0] = 0.007813
<S1|S0> = 0.010872
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 349.69 sec.
9 -3505.0916994333 2.14E-04
Det[Ca.S.C0] = 0.984519
Det[Cb.S.C0] = 0.008355
<S1|S0> = 0.011633
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.93 sec.
Timing for roks_build(): 348.59 sec.
10 -3505.0945412366 6.19E-05
Det[Ca.S.C0] = 0.984593
Det[Cb.S.C0] = 0.008613
<S1|S0> = 0.011993
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.93 sec.
Timing for roks_build(): 348.22 sec.
11 -3505.0947841246 1.54E-05
Det[Ca.S.C0] = -0.984601
Det[Cb.S.C0] = -0.008776
<S1|S0> = 0.012220
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.93 sec.
Timing for roks_build(): 347.81 sec.
12 -3505.0948024863 2.22E-06
Det[Ca.S.C0] = -0.984597
Det[Cb.S.C0] = -0.008908
<S1|S0> = 0.012404
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.93 sec.
Timing for roks_build(): 349.90 sec.
13 -3505.0948046481 1.49E-06
Det[Ca.S.C0] = -0.984592
Det[Cb.S.C0] = -0.009072
<S1|S0> = 0.012632
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.93 sec.
Timing for roks_build(): 349.12 sec.
14 -3505.0948067508 1.32E-06
Det[Ca.S.C0] = 0.984585
Det[Cb.S.C0] = 0.009397
<S1|S0> = 0.013085
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.95 sec.
Timing for roks_build(): 349.83 sec.
15 -3505.0948090291 1.17E-06
Det[Ca.S.C0] = -0.984573
Det[Cb.S.C0] = -0.010028
<S1|S0> = 0.013963
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.94 sec.
Timing for roks_build(): 349.03 sec.
16 -3505.0948100492 1.04E-06
Det[Ca.S.C0] = -0.984556
Det[Cb.S.C0] = -0.010862
<S1|S0> = 0.015124
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.93 sec.
Timing for roks_build(): 348.79 sec.
17 -3505.0948052955 8.77E-07
Det[Ca.S.C0] = -0.984532
Det[Cb.S.C0] = -0.011941
<S1|S0> = 0.016625
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.88 sec.
Timing for roks_build(): 349.13 sec.
18 -3505.0947908689 6.96E-07
Det[Ca.S.C0] = -0.984505
Det[Cb.S.C0] = -0.012932
<S1|S0> = 0.018005
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.87 sec.
Timing for roks_build(): 347.66 sec.
19 -3505.0947703687 5.13E-07
Det[Ca.S.C0] = -0.984481
Det[Cb.S.C0] = -0.013673
<S1|S0> = 0.019036
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.91 sec.
Timing for roks_build(): 348.02 sec.
20 -3505.0947509411 3.70E-07
Det[Ca.S.C0] = -0.984466
Det[Cb.S.C0] = -0.014069
<S1|S0> = 0.019587
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 347.81 sec.
21 -3505.0947394175 2.76E-07
Det[Ca.S.C0] = -0.984456
Det[Cb.S.C0] = -0.014326
<S1|S0> = 0.019945
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.91 sec.
Timing for roks_build(): 348.50 sec.
22 -3505.0947315107 2.04E-07
Det[Ca.S.C0] = 0.984452
Det[Cb.S.C0] = 0.014459
<S1|S0> = 0.020131
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.92 sec.
Timing for roks_build(): 349.20 sec.
23 -3505.0947270909 1.66E-07
Det[Ca.S.C0] = 0.984449
Det[Cb.S.C0] = 0.014548
<S1|S0> = 0.020254
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 347.91 sec.
24 -3505.0947243592 1.79E-07
Det[Ca.S.C0] = 0.984446
Det[Cb.S.C0] = 0.014630
<S1|S0> = 0.020369
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.93 sec.
Timing for roks_build(): 348.17 sec.
25 -3505.0947216356 9.30E-08
Det[Ca.S.C0] = 0.984445
Det[Cb.S.C0] = 0.014687
<S1|S0> = 0.020448
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.89 sec.
Timing for roks_build(): 348.73 sec.
26 -3505.0947196915 8.32E-08
Det[Ca.S.C0] = 0.984443
Det[Cb.S.C0] = 0.014734
<S1|S0> = 0.020512
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.91 sec.
Timing for roks_build(): 349.04 sec.
27 -3505.0947180776 5.98E-08
Det[Ca.S.C0] = 0.984443
Det[Cb.S.C0] = 0.014750
<S1|S0> = 0.020535
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.88 sec.
Timing for roks_build(): 348.78 sec.
28 -3505.0947174973 3.46E-08
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014760
<S1|S0> = 0.020549
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.91 sec.
Timing for roks_build(): 348.29 sec.
29 -3505.0947171109 2.37E-08
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014767
<S1|S0> = 0.020558
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 348.00 sec.
30 -3505.0947168584 1.60E-08
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014768
<S1|S0> = 0.020560
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 348.52 sec.
31 -3505.0947168073 7.73E-09
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014766
<S1|S0> = 0.020557
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.69 sec.
Timing for roks_build(): 349.31 sec.
32 -3505.0947168674 3.58E-09
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014766
<S1|S0> = 0.020557
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 348.58 sec.
33 -3505.0947168716 2.22E-09
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014765
<S1|S0> = 0.020556
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 349.67 sec.
34 -3505.0947168959 1.35E-09
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014765
<S1|S0> = 0.020556
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.90 sec.
Timing for roks_build(): 347.50 sec.
35 -3505.0947168974 1.09E-09
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014765
<S1|S0> = 0.020556
Building long-range K (w = 3.300e-01)
Building long-range K (w = 3.300e-01)
Timing for roks_project_singlet(): 3.84 sec.
Timing for roks_build(): 347.10 sec.
36 -3505.0947169094 9.12E-10 Convergence criterion met
---------------------------------------
One-Electron Energy = -12934.6127500560
Total Coulomb Energy = 5629.0257291864
Alpha Exchange Energy = -35.3734631443
Beta Exchange Energy = -36.1129889774
DFT Exchange Energy = -199.3215623933
DFT Correlation Energy = -10.8076683049
Nuclear Repu. Energy = 4082.1079867803
Nuclear Attr. Energy = -16428.0189075641
Kinetic Energy = 3493.4061575081
Det[Ca.S.C0] = 0.984442
Det[Cb.S.C0] = 0.014764
<S1|S0> = 0.020555
SCF time: CPU 12699.87 s wall 626.31 s
That overlap does not look bad at all. Is there any particular reason you’re using GEN_SCFMAN = FALSE? You could try with GEN_SCFMAN and use different SCF solvers (DIIS + MOM, SGM, I even use GDM some times for these…) Also, are you sure the guess you’re providing is correct? In other words, are you sure that the HOMO and LUMO from the guess look like the NTOs of the state you’re trying to target?
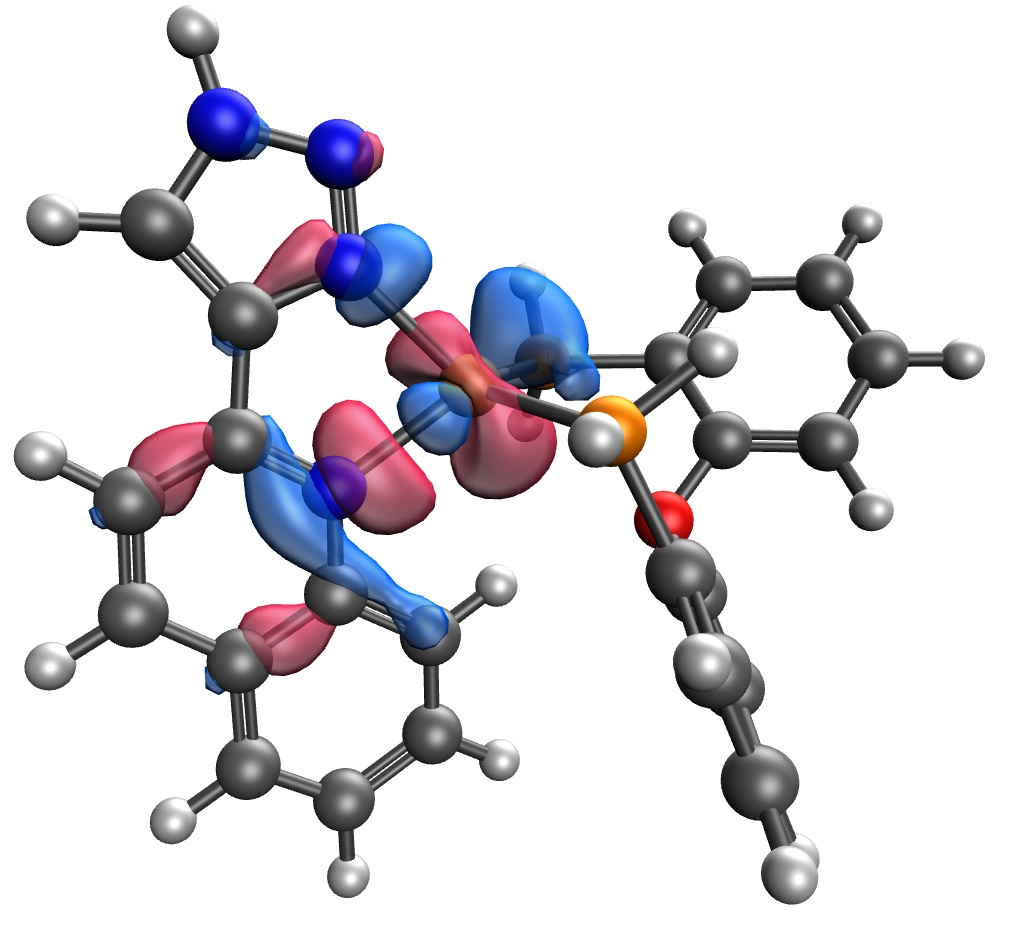
I ran a test on a small basis with GEN_SCFMAN = TRUE and using GDM as the SCF solver, plus a small trick I implemented for ROKS (will be out in 6.2). I cant show you a difference density because I dont know how to plot those but the two open-shell orbitals of the ROKS calculations seem consistent with the difference density you show for TDDFT. Isovalues of 0.100.
I finally got back to the ROKS calculations.
Thank you for your advice! Changing the SCF solver to GDM and GEN_SCFMAN to true worked, the difference density looks similarly to TD-DFT one.
I was using gen_scfman = false because in the first job I was generating ground state orbitals in a basis set that’s not built in in Q-Chem (DZP).
However, I have a couple more questions.
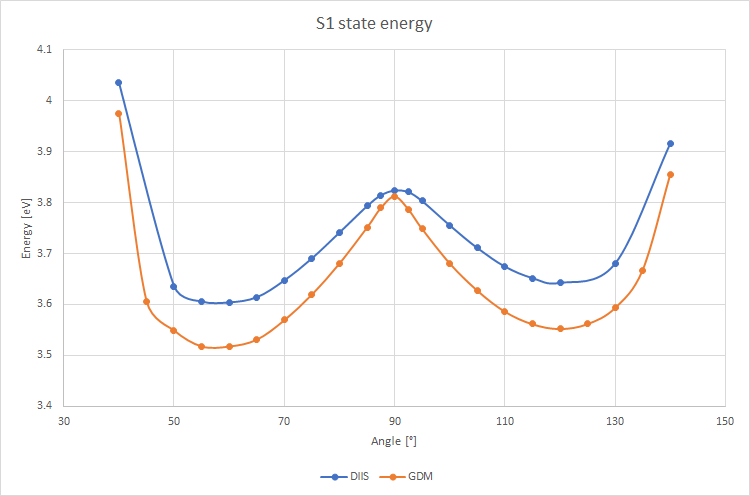
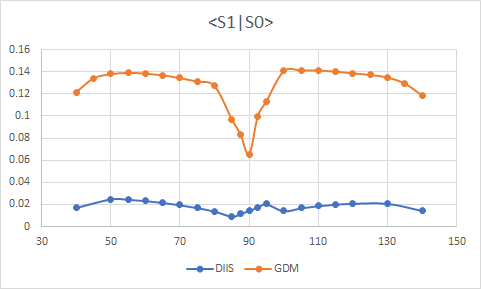
I am doing a PES scan of S1 state along the rotation of N-Cu-N and P-Cu-P planes. Using the two SCF solvers I got the following picture:
Glad it worked. The unfortunate answer is yes. Unlike ROHF for high-spin open-shell systems, the ROKS energy is not invariant to rotations among the open shells. As a consequence the orbital space may have several local minima that look wonky (weird difference densities, large overlaps with the ground state, bad energies…) and different solvers will sometimes land you in different ones. In general, I like the ROKS solutions that have the smallest overlap with the ground state and that “look” correct (MOs / orbital differences make sense). In my experience they have often gone hand-in-hand but yours is the first time I see when a smaller overlap (DIIS solution) produces wonky orbitals. Could it be that the wonky DIIS solution is a legit solution that just doesnt look like the one you’re interested on? (i.e. a nearby excited state)
As I mentioned, I implemented a trick that supressess the open-shell mixing in ROKS and it does wonders with GDM and SGM. So, if you provide it a guess with sensible open-shell orbitals (ground state orbitals) it’ll try to prevent the open-shells from getting scrambled up and producing weird results. It is an unpublished idea since I didnt have the time to write it up so use at your own discretion! It’ll be out in 6.2 so you can try it then. I’d also be happy to run a test one for you to see what happens but I’d need the exact input file.