Dear Sir or Madam,
Here is my question ,we use bend to adjust H-O-H bond , but it’s support to 180°.This is information of output file.

Linear (or near-linear) angles can lead to undefined behavior because the internal-to-cartesian transformation is multi-valued. Perhaps better to use distance constraints. If you use both O-H and H-H distance, you can accomplish the same objective this way.
As @jherbert states, angles which are near linear ( Angle >= 165) are considered linear angles (180) within the code to help prevent issues which may crop up when an angle is optimized to a linear angle. Depending on the structure, constraining the bonds may be the only option.
If you provide the input I may be better able to provide insight into the issue and possible solutions.
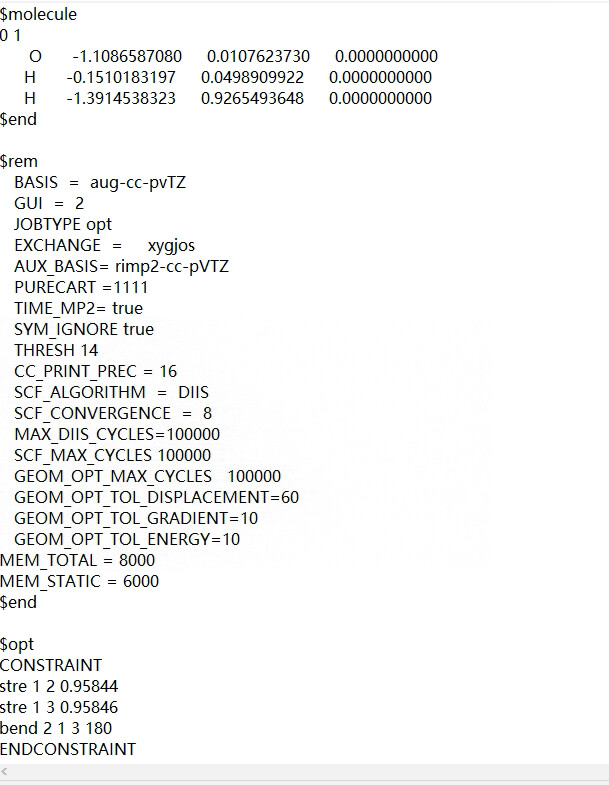
Thank for you reply.As you said,we want to adjust the H-O-H bond , bend constrain will be the most convenient.There is the inputfile.
So it isn’t obvious to me right away but can you explain what you are trying to optimize?
The problem I have is that for example if you take 3 atom linear molecule where the only constraint is the angle to 180 degrees (A-B-C), the only 2 optimizable coordinates are the bond lengths (A-B, B-C). Continuing with this example if the two bond constraints are added to the existing angle constraint, there are no optimizable coordinates. I would then claim that the structure is already at the minimum given the constraints.
Therefore with your input file, I would claim you wouldn’t need to do a optimization job, but just use the input structure for any further analysis (Energy, Gradient, Frequency, Excited State, etc.) as with the given constraints you have the minimized structure.
I’m sorry I didn’t make myself clear, but in this calculation, we basically want to change the Angle of the H-O-H bond, and we can go on without limiting the length of the bond. However, the difficulty now is that the H-O-H Angle does not support up to 180°, which would make our calculation meaningless.
Ok If you want to change/optimize the H-O-H angle all you need to do is constrain the two bonds.
$opt
CONSTRAINT
stre 1 2 0.95844
stre 1 3 0.95846
ENDCONSTRAINT
$end
I’m sorry, Sir, but I may not have made myself clear. The difficulty I encountered was that neither limitation optimization nor potential energy surface scanning could be supported to 180° with bend.This is error information in outfile.
Angle Constraint 1 is Greater than Near-Linear Threshold 165.0
Depend on use O-H and H-H distance,couldn’t achevie the purpose.The difficulty I encountered was that neither limitation optimization nor potential energy surface scanning could be supported to 180° with bend.This is error information in outfile.
Angle Constraint 1 is Greater than Near-Linear Threshold 165.0
So,we want to know the angles whether Values in degrees, 0 ≤ value ≤ 180 is correctly use or not.
Look for your reply.
If all you need is a 1d potential scan along the bond angle, with fixed bond lengths, then why not simply generate a sequence of input files and do the scan manually, as single-point calculations?
Hello xrq,
Cartesian coordinate is less efficient than the default, internal. However, you can use GEOM_OPT_COORDS = 0 to avoid the restriction. (9.2.2 OptimizeJob Control⣠9.2 Geometry Optimization Job Controls ⣠Chapter 9 Exploring Potential Energy Surfaces: Searches for Critical Points and Molecular Dynamics ⣠Q-Chem 6.0 Userâs Manual)
Kuan-Yu