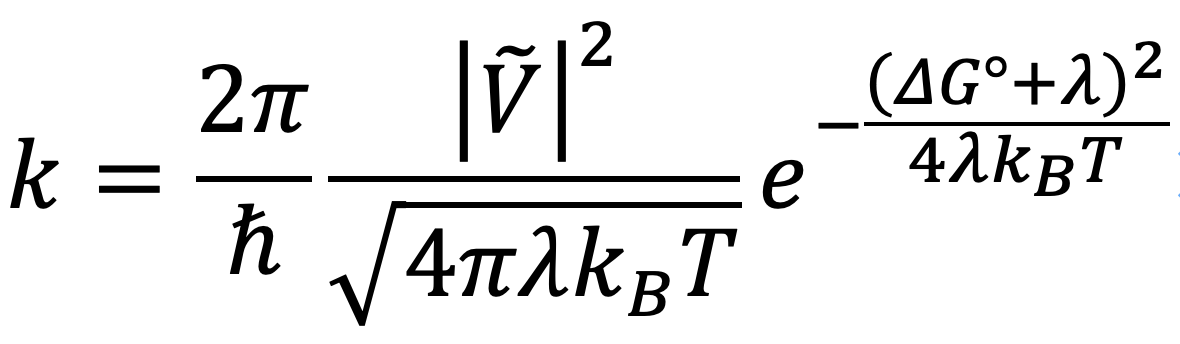Hello,
I am new to Q-Chem. I found that the software can compute the V-coupling value for energy transfer, but I am not sure whether my input is correct or how to interpret the output. I would greatly appreciate any advice.
Specifically, I am interested in the V-coupling associated with energy transfer between a photosensitizer in its T₁ state and ground-state oxygen (³O₂). This corresponds to Dexter energy transfer, relevant to photodynamic therapy:
(T₁)Ps + ³O₂ → (S₀)Ps + ¹O₂.
My input file is:
$rem
METHOD TPSSh
BASIS def2-TZVP
DFT_D D4
SOLVENT_METHOD SMD
STS_FSD TRUE
CIS_N_ROOTS 10
CIS_SINGLETS true
CIS_TRIPLETS true
STS_DONOR 1-30
STS_ACCEPTOR 31-32
$end
$smx
solvent water
$end
$comment
EET
$end
$molecule
0 3
O -2.296744 -2.636037 -0.968211
O 0.206036 -2.461183 -0.752196
O -0.302321 2.762782 0.729913
O 2.704473 -2.257589 -0.588512
O 4.604231 1.745639 0.881824
C -1.334952 0.746758 0.046143
C -1.192059 -0.597776 -0.324505
C 1.225901 -0.419771 -0.111092
C 1.101576 0.926772 0.252759
C 0.083910 -1.242222 -0.419295
C -0.190619 1.581981 0.368580
C -2.605848 1.330528 0.127365
C -2.366042 -1.363398 -0.616727
C -3.737982 0.589971 -0.162551
C 2.546362 -1.004797 -0.220463
C 2.234332 1.646628 0.646742
C -3.606182 -0.746016 -0.530377
C 3.508965 1.077929 0.542425
C 3.651722 -0.233973 0.078403
C -5.093994 1.212293 -0.087685
H -2.663393 2.368238 0.418378
H 2.100911 2.654297 1.012808
H -4.473697 -1.347256 -0.753019
H 4.632226 -0.675591 0.012222
H -5.545497 1.222949 -1.077890
H -5.736109 0.625488 0.565066
H -5.035232 2.229329 0.285655
H -1.342201 -2.925586 -0.977466
H 1.806313 -2.674694 -0.744708
H 4.394135 2.641501 1.196817
O 2.041183 0.109198 2.744029
O 1.519313 -0.935044 2.262480
$end
My uncertainties are as follows:
- I am not sure whether to use STS_FSD or STS_FED. The manual notes that FED excludes the ground state from the analysis, but I need the ground state included for ³O₂.
- I specified the total system as 0 3 without explicitly defining fragments (e.g., 0 1 Ps, 0 3 O₂, which also sums to 0 3). I am not confident that this setup correctly enforces the photosensitizer in T₁ and oxygen in its triplet ground state.
The relevant portion of my output is:
FSD ELECTRONIC-COUPLING CALCULATION
Fragment Spins of Ground State
-----------------------------------------------------
State S(D) S(A) dS
-----------------------------------------------------
0 0.149564 1.850436 -1.700872
-----------------------------------------------------
Within CIS/TDA Excited States:
Fragment Spins of Excited State
-----------------------------------------------------
State S(D) S(A) dS
-----------------------------------------------------
1 0.258255 1.741745 -1.483491 ( 0.217381)
2 0.235453 1.764547 -1.529093 ( 0.171778)
3 0.936233 1.063767 -0.127534 ( 1.573338)
4 0.258098 1.741902 -1.483804 ( 0.217067)
5 0.235280 1.764720 -1.529441 ( 0.171431)
6 0.575434 1.424566 -0.849131 ( 0.851740)
7 0.687242 1.312758 -0.625516 ( 1.075355)
8 0.962554 1.037446 -0.074891 ( 1.625980)
9 0.822590 1.177410 -0.354819 ( 1.346053)
10 0.815321 1.184679 -0.369359 ( 1.331513)
-----------------------------------------------------
Fragment Spins of Transition Densities and
FSD Couplings Between Ground and Excited States
--------------------------------------------------------------------------------
States S12(D) S12(A) dS12 Coupling(eV) FSD-dS1 FSD-dS2
--------------------------------------------------------------------------------
0 1 -0.018143 0.018143 -0.036285 0.2746709 -1.706768 -1.477594
0 2 -0.006089 0.006089 -0.012177 0.1408457 -1.701731 -1.528235
0 3 0.004742 -0.004742 0.009484 -0.01249373 -1.700929 -0.127476
0 4 -0.001072 0.001072 -0.002144 0.02320875 -1.700893 -1.483783
0 5 0.026552 -0.026552 0.053104 -0.6388357 -1.715989 -1.514324
0 6 0.076665 -0.076665 0.153329 -0.4153414 -1.727633 -0.822370
0 7 -0.058739 0.058739 -0.117478 0.265195 -1.713556 -0.612832
0 8 0.012239 -0.012239 0.024478 -0.03862859 -1.701240 -0.074523
0 9 -0.092387 0.092387 -0.184775 0.3443387 -1.725775 -0.329915
0 10 0.041865 -0.041865 0.083730 -0.1654571 -1.706116 -0.364114
--------------------------------------------------------------------------------
Fragment Spins of Transition Densities and
FSD Couplings Between Excited States
--------------------------------------------------------------------------------
States S12(D) S12(A) dS12 Coupling(eV) FSD-dS1 FSD-dS2
--------------------------------------------------------------------------------
1 2 -0.022584 0.022584 -0.045168 -0.1213628 -1.455695 -1.556889
1 3 0.091768 -0.091768 0.183535 -0.04416684 -1.507894 -0.103130
1 4 -0.027672 0.027672 -0.055344 -0.3078439 -1.428304 -1.538991
1 5 0.033478 -0.033478 0.066956 0.3268944 -1.435677 -1.577254
1 6 0.058255 -0.058255 0.116509 -0.1236803 -1.504212 -0.828410
1 7 -0.053195 0.053195 -0.106390 0.09026335 -1.496486 -0.612521
1 8 -0.009792 0.009792 -0.019585 0.01156735 -1.483763 -0.074619
1 9 0.096739 -0.096739 0.193478 -0.1405041 -1.515736 -0.322574
1 10 0.004786 -0.004786 0.009571 -0.007877437 -1.483573 -0.369276
2 3 0.146756 -0.146756 0.293513 -0.01277733 -1.588078 -0.068549
2 4 0.010671 -0.010671 0.021343 -0.1178988 -1.537566 -1.475332
2 5 -0.001250 0.001250 -0.002499 -0.2091495 -1.526762 -1.531772
2 6 0.011266 -0.011266 0.022532 -0.0147303 -1.529839 -0.848385
2 7 0.089148 -0.089148 0.178297 -0.08774905 -1.563003 -0.591607
2 8 -0.006464 0.006464 -0.012928 0.004981156 -1.529208 -0.074777
2 9 0.135311 -0.135311 0.270622 -0.1244424 -1.588459 -0.295453
2 10 0.015156 -0.015156 0.030313 -0.01684149 -1.529885 -0.368567
3 4 -0.000938 0.000938 -0.001875 -0.0003839019 -0.127531 -1.483807
3 5 0.036994 -0.036994 0.073989 0.01853574 -0.123639 -1.533335
3 6 0.096087 -0.096087 0.192173 0.08916712 -0.079546 -0.897119
3 7 -0.061024 0.061024 -0.122048 -0.09065213 -0.099230 -0.653820
3 8 0.014851 -0.014851 0.029703 -0.1849485 -0.140900 -0.061526
3 9 0.021478 -0.021478 0.042956 0.09341853 -0.119686 -0.362666
3 10 -0.007445 0.007445 -0.014890 -0.03538849 -0.126620 -0.370272
4 5 -0.052060 0.052060 -0.104119 -0.03688322 -1.400032 -1.613213
4 6 -0.065880 0.065880 -0.131759 0.01950065 -1.510071 -0.822865
4 7 -0.085399 0.085399 -0.170797 0.02482829 -1.516544 -0.592777
4 8 0.248861 -0.248861 0.497721 -0.06249048 -1.641894 0.083198
4 9 0.061526 -0.061526 0.123053 -0.02670689 -1.497061 -0.341563
4 10 -0.041143 0.041143 -0.082286 0.02201707 -1.489847 -0.363316
5 6 0.132030 -0.132030 0.264061 -0.00803021 -1.619906 -0.758666
5 7 0.135449 -0.135449 0.270898 -0.01510652 -1.604409 -0.550548
5 8 -0.066649 0.066649 -0.133297 0.01271638 -1.541555 -0.062777
5 9 -0.068416 0.068416 -0.136832 0.01988441 -1.545170 -0.339090
5 10 -0.005675 0.005675 -0.011351 0.002209949 -1.529552 -0.369248
6 7 0.376985 -0.376985 0.753971 -0.01611253 -1.499539 0.024892
6 8 0.097690 -0.097690 0.195380 -0.02588126 -0.895641 -0.028381
6 9 -0.169734 0.169734 -0.339469 0.06025904 -1.021886 -0.182064
6 10 0.001801 -0.001801 0.003601 -0.001498964 -0.849158 -0.369332
7 8 -0.023385 0.023385 -0.046769 0.006892199 -0.629461 -0.070947
7 9 0.175978 -0.175978 0.351956 -0.05436798 -0.867252 -0.113083
7 10 -0.012383 0.012383 -0.024766 0.01586526 -0.627889 -0.366986
8 9 0.008235 -0.008235 0.016471 0.001998137 -0.073926 -0.355785
8 10 0.069647 -0.069647 0.139294 0.02915074 -0.019442 -0.424809
9 10 0.042201 -0.042201 0.084401 0.02522624 -0.277375 -0.446803
--------------------------------------------------------------------------------
END OF FSD CALCULATION
My interpretation is that State 0 corresponds to S₀ of the photosensitizer plus T₁ of O₂. However, none of the excited states seem to represent T₁ of the photosensitizer, since none of the fragment spin values approach S = 2.
How should I correctly set up the calculation so that the photosensitizer T₁ and O₂ (³O₂) are properly captured?
THANKS!