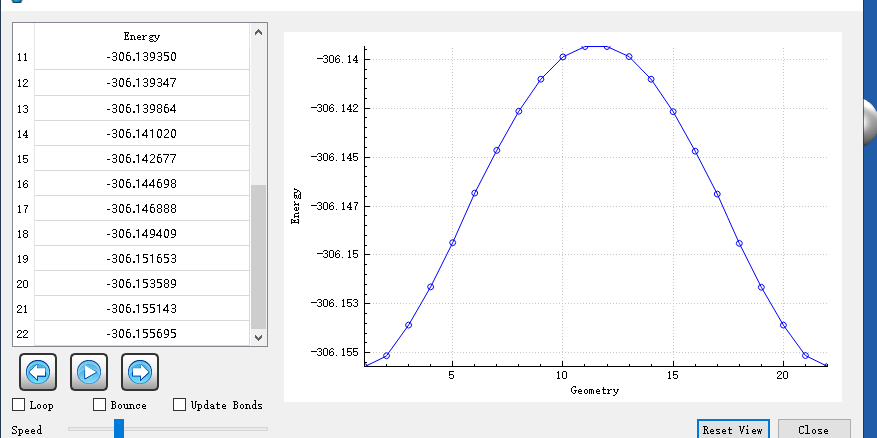
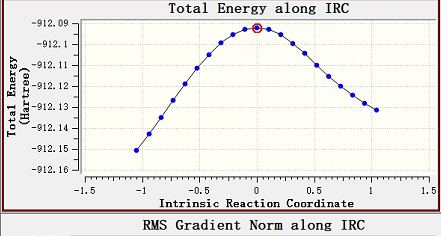
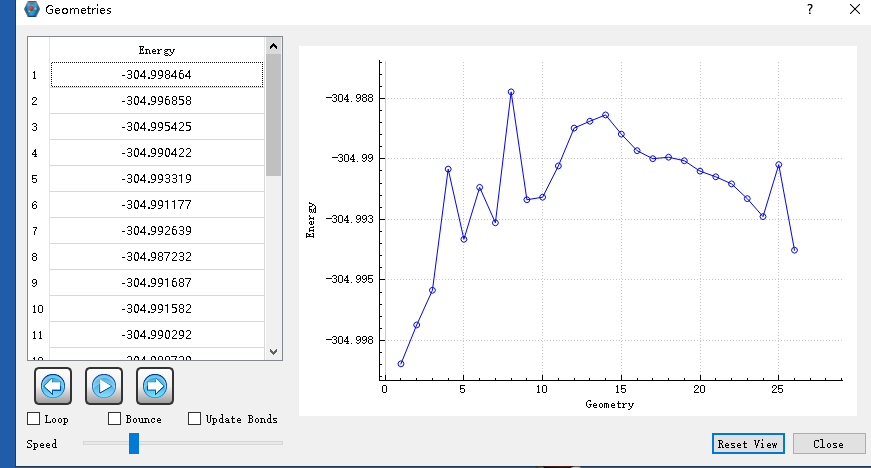
But we have encountered a new problem. We would like to get the total energy along the IRC direction, but we cannot find the values of the intrinsic reaction coordinate. And through I-Qmol only GEOMETRY vs Energy figure can be observed. Waiting for your reply.And How is the IRC figure obtained?
Thank you for your help. We calculate the transition state from reactant and product structures and added “JOBTYPE rpath”. The input file command is as follows:
$rem
JOBTYPE fsm
METHOD pbe0
DFT_D = D3_BJ
BASIS aug-cc-pVTZ
FSM_NGRAD 3
FSM_NNODE 18
FSM_MODE 2
FSM_OPT_MODE 2
SYMMETRY false
SYM_IGNORE true
$end
However, where do we look for the correct IRC figure?in .out/Vfile.txt/stringfile.txt/perp_grad_file.txt
At present, i observe the figure by IQMOL is ambiguous and not smooth.Waiting for your reply.
It’s unlikely than anyone can diagnose this problem without the full input file (including coordinates). Furthermore, it is helpful to provide a minimalist example that runs quickly (small molecule and small basis sets). Along those lines, I don’t know why anyone would optimize geometries at PBE0+D3 level using aug-cc-pVTZ basis. Shouldn’t be that sensitive to basis set, 6-31G(d) or 6-31+G(d) is probably just as good and much faster.